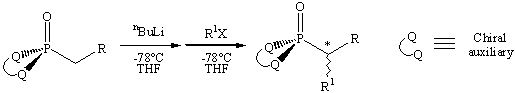AmericanChemicalSociety.com
Reports: AC3 47563-AC3: New Chiral Solid Catalysts for Oxidation Reactions
Asymmetric catalytic reactions are some of the most interesting and fundamentally important processes in chemistry. Several asymmetric processes are used in industry and these include key intermediate steps in the synthesis of pheromones, Vitamin E, aspartame, and several key therapeutic drugs. Although most commercially significant asymmetric transformations use homogeneous catalysis, research in the past two decades has intensified in the area of asymmetric heterogeneous catalysis. The thrust behind this interest is to develop recyclable catalysts that reduce the costs of unrecoverable metals and ligands, that are easy to separate from product mixtures, and that improve the ‘green-ness' of the industrial process.
Heterogeneous catalysts are commonly prepared by ‘heterogenizing' existing homogeneous catalysts or by altering catalytic materials with chiral modifiers. Common ‘heterogenization' techniques include tethering catalysts to insoluble supports, occluding catalysts in organic or inorganic polymeric supports, and using biphasic mixtures to separate the catalyst from the substrates and products. A slightly different approach is to synthesize a catalytically active insoluble polymer from enantiopure molecular building blocks. In this approach, all of the metal sites of the surface would be catalytically active and the use of chiral ligand building blocks would result in the formation of a chiral surface.
We are exploring the synthesis of new chiral heterogeneous catalysts by treating vanadium starting materials with phosphonic acids. Achiral vanadium phosphonates are known that adopt a variety of structures, including soluble cage and cluster compounds, 1-dimensional chain structures, 2-dimensional layered structures, and 3-dimensional polymeric structures. To date, no reports of the synthesis of vanadium phosphonates with enantiopure phosphonic acids has been published.
We have prepared enantiopure (1-phenylalkyl)phosphonic acids by using 1,1'-binaphthyl-2,2'-diol, (BINOL), as a chiral auxiliary (Figure 1). The alkylation of lithium-stabilized alkyl phosphonates is known to proceeds stereoselectively if the phosphonate is chelated with a chiral auxiliary. This lithium-stabilized alkylation reaction has been studied extensively and the most common chiral auxiliaries are trans-N,N'-dialkyl-1,2-diaminoalkanes, (-) ephedrine, and other substituted N-alkyl-b-amino alcohols. Using these auxiliaries, chiral phosphonates are produced in high yields (> 90%) and with excellent diastereoselectivities (98 : 2). However, these chiral auxiliaries can be expensive, can take multiple steps to prepare, and can hydrolyze under humid conditions.
Figure 1: Asymmetric alkylation of lithium-stabilized phosphonates.
To determine if
1,1'-binaphthyl-2,2'-diol, BINOL, could also serve as a chiral
auxiliary, (S(-)BINOL)P(O)CH2Ph was prepared by treatment of
Cl2P(O)CH2Ph with S(‑)BINOL in the presence
of two equivalents of triethylamine. Resolved BINOL is commercially available,
relatively inexpensive, and C2-symmetric, and (BINOL)P(O)CH2Ph was found to be stable under humid
conditions.
We
find that deprotonation of enantiopure
[S(-)(1,1'-binaphthyl-2,2'-dioxo)benzylphosphonate,
[S(-)BINOL]P(O)CH2Ph), followed by addition of an alkyl halide, RX,
where R = methyl, ethyl, allyl, or benzyl, at -72 °C affords diastereoenriched
(1-phenylalkyl)phosphonates (S(-)BINOL)P(O)CHRPh in good yields (up to 94%) and good diastereoselectivities (up to 91 :9). After the diastereomers
were separated by flash column chromatography and the chiral
auxiliary was removed under basic conditions, enantiopure
(1-phenylalkyl)phosphonic
acids (HO)2P(O)(CHRPh) can be isolated. Treatment of
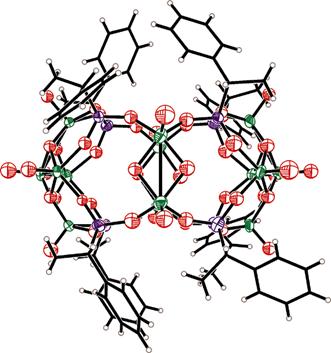
vanadium starting materials with chiral and achiral phosphonic acids and phosphonates provided the soluble [BuPPh3]4[(V2O2(OH)2)6(O3PR)8]
R = CH(CH2Ph)Pr and CH(Et)Pr, [(VO)6(O3PR)8
Figure 2.
Molecular structure of [(V14O22)(OH)4(O3PCH(Me)Ph-S)8]6- Tests for catalytic activity in the asymmetric epoxidation and other oxidation reactions are currently
underway.