AmericanChemicalSociety.com
Reports: G1 48346-G1: Addition-cycloisomerization of Propargylcyanamides; Efficient Access to the 2-aminoimidazole Core
Synthetic platform: Given the
range of structural diversity in these natural products in respect to ring
size, oxidation state and substitution we aimed to develop a unified synthetic
platform to access these skeletons. In pursuance of our 1) provide access to
predictable and controlled substitution patterns in short order, 2) deliver these diverse hydrogen bond topologies, and 3)
permit
ring oxidation state adjustments within these cyclic or polyclic
structures. We
felt that the addition of a guanidine N-H bond across a C-C p-system would represent a powerful
tool for the preparation of these challenging heterocyles.
The development of amine-alkene/alkyne hydroamination
chemistry has provided an invaluable tool for the construction of nitrogen
based heterocycles. The extension of this methodology
to guanidines, however, remains largely
underdeveloped. In order to manipulate the oxidation states of these
heterocyclic cores we anticipated that this is best done by reduction, as
oxidations of nitrogen rich heterocycles are usually
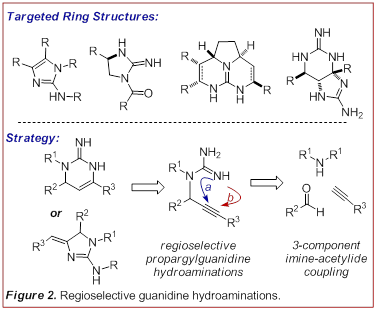
problematic. This has led us to study the addition of guanidine N-H bonds across
alkynes, thus providing access to cyclic structures at an elevated oxidation
state (Figure 2). This disconnection also permits the rapid construction of
diverse skeletal precursors via an imine-acetylide
3-component coupling. To this end we examined the utility of an addition-hydroamination-isomerization sequence (Figure 3). (Angew. Chem. Int. Ed. 2009, 48, 3116-3120). This strategy provided access to a variety of
highly substituted 2-aminoimidazoles in just 3 steps and was generally high
yielding. Further, the introduction of a removable R group permitted the
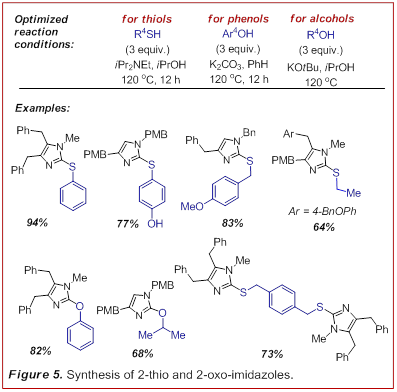
preparation of any desired hydrogen bond donor-acceptor topology. We have also been able to adapt this
reaction sequence to the addition of thiols, phenols
and alcohols (Figure 5, J. Org. Chem.
2010, 75, 261-264). Consistent with what we had
learned from the addition of amines to the cyanamides,
this chemistry requires a fine balance of nucleophilicity
and basicity to effect addition to the cyanamide
without formation of the allenyl-cyanamide which
readily decomposes. This manifold presents an interesting polarity reversal for
the preparation of these heterocycles which are
usually constructed by the addition of an intact thiourea
or urea to an electrophile. The major limitation of this methodology was that the
addition of a nucleophile to the cyanamide was rate
limiting and
 Our
research program ultimately requires synthetic chemistry to study the
biological functions of structurally unique natural products with significant
biomedical relevance. We have a continued interest in the synthesis and biology
of guanidine containing natural products, due to the importance of this motif
in the molecular recognition of carboxylate, sulfate and phosphate groups in
biological macromolecules (Curr. Bioact. Cmpds. 2009, 5,
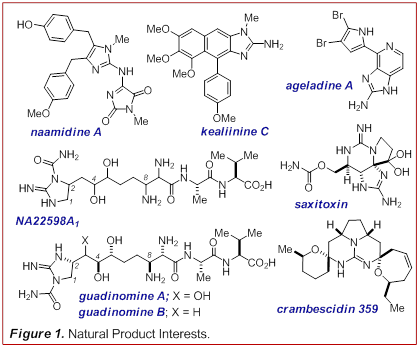
39-78). We are interested in the Leucetta alkaloids of which naamidine
A and kealinine C are representatives (Figure 1).
These compounds display a diverse range of biological activities based on a
relatively conserved structural core. In particular we are interested in the
unique ability of naamidine A to modulate EGF
dependent DNA synthesis. Ultimately this hyper stimulation leads to activation
of caspase-dependent apoptosis. While allosteric
kinase inhibitors are well characterized, the roles of allosteric agonists are
relatively unknown. Synthetic access to these compounds will provide probes to
further delineate the important activity of naamidine
A. NA22598A1 or the guadinomines also
represent an important arena for target identification and mechanism of action
studies. The only difference between
these compounds is the purported position of the carbamoyl
group on the guanidine (either N1 or N2). Since both natural
products are isolated from Streptomyces sp. It is our contention that
these are the same compound and that erroneous HMBC data in the structure
elucidation of NA22598A1 led to an incorrect structural interpretation. Initial
spectroscopic investigations in our laboratory on model systems indicate that
this is indeed the case. NA22598A1 was identified as a selective inhibitor of
anchorage independent growth (AIG), suggesting a potential lead for small
molecules that can selectively inhibit tumor metathesis. The guadinomines were identified as agents capable of
inhibiting the Type 3 Secretion System (T3SS). If it is true that these are the
same compounds, it would suggest a unique and unknown biochemical link between
these two biological processes. We hypothesize that both of these processes may
be modulated by the activity of matrixmetalloproteinases
and that these natural products are thus inhibitors of MMPs. Further we suggest
that the carbamoyl guanidine is a unique motif
capable of divalent metal chelation and thus provides a hypothesis for the
mechanism of inhibition of these Zn(II)dependent metalloenzymes. If this is indeed the case, it would
represent an important advance for the design of MMP inhibitors. Ageladine A is also an MMP inhibitor but its exact mode of
inhibition has not been characterized with atomic resolution. Other marine
metabolites with guanidines serve as synthetic
inspiration such as saxitoxin, the crambescidins and the batzelladines.
Our
research program ultimately requires synthetic chemistry to study the
biological functions of structurally unique natural products with significant
biomedical relevance. We have a continued interest in the synthesis and biology
of guanidine containing natural products, due to the importance of this motif
in the molecular recognition of carboxylate, sulfate and phosphate groups in
biological macromolecules (Curr. Bioact. Cmpds. 2009, 5,
39-78). We are interested in the Leucetta alkaloids of which naamidine
A and kealinine C are representatives (Figure 1).
These compounds display a diverse range of biological activities based on a
relatively conserved structural core. In particular we are interested in the
unique ability of naamidine A to modulate EGF
dependent DNA synthesis. Ultimately this hyper stimulation leads to activation
of caspase-dependent apoptosis. While allosteric
kinase inhibitors are well characterized, the roles of allosteric agonists are
relatively unknown. Synthetic access to these compounds will provide probes to
further delineate the important activity of naamidine
A. NA22598A1 or the guadinomines also
represent an important arena for target identification and mechanism of action
studies. The only difference between
these compounds is the purported position of the carbamoyl
group on the guanidine (either N1 or N2). Since both natural
products are isolated from Streptomyces sp. It is our contention that
these are the same compound and that erroneous HMBC data in the structure
elucidation of NA22598A1 led to an incorrect structural interpretation. Initial
spectroscopic investigations in our laboratory on model systems indicate that
this is indeed the case. NA22598A1 was identified as a selective inhibitor of
anchorage independent growth (AIG), suggesting a potential lead for small
molecules that can selectively inhibit tumor metathesis. The guadinomines were identified as agents capable of
inhibiting the Type 3 Secretion System (T3SS). If it is true that these are the
same compounds, it would suggest a unique and unknown biochemical link between
these two biological processes. We hypothesize that both of these processes may
be modulated by the activity of matrixmetalloproteinases
and that these natural products are thus inhibitors of MMPs. Further we suggest
that the carbamoyl guanidine is a unique motif
capable of divalent metal chelation and thus provides a hypothesis for the
mechanism of inhibition of these Zn(II)dependent metalloenzymes. If this is indeed the case, it would
represent an important advance for the design of MMP inhibitors. Ageladine A is also an MMP inhibitor but its exact mode of
inhibition has not been characterized with atomic resolution. Other marine
metabolites with guanidines serve as synthetic
inspiration such as saxitoxin, the crambescidins and the batzelladines.
 interests,
a unified approach must:
interests,
a unified approach must: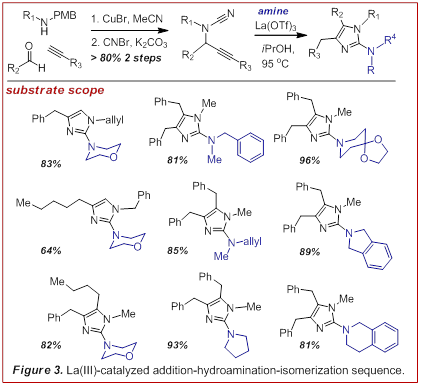
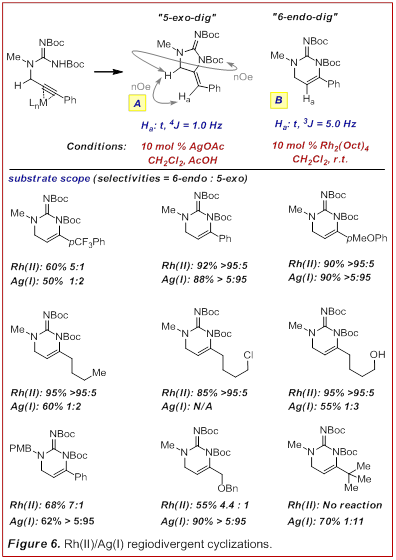 required forcing temperatures (>95 oC). Under these conditions only the stereo-electronically
favored 5-exo-dig cyclization was observed followed by isomerization. Thus we
set out to develop a more mild cyclization procedure
that would allow us to A) selectively generate the 5-exo-dig or
the 6-endo-dig product and B) maintain the fidelity of the alkene
position so that it can be used for further functionalization. Under the
simplistic assumption that a p-Lewis acid might
trigger the hydroamination process, a series of metal
salts were evaluated for their ability to affect the cyclization of an intact N,N-diboc-propargylguanidine
(Figure 6, Angew. Chem. Int. Ed.
2011, 50, 684-687). In
short we found that Ag(I) catalysts were optimum for
the formation of the formal 5-exo-dig product. We also discovered a
unique role of dirhodium carboxylates to promote the
selective formation of the formal 6-endo-dig product. Of particular
note is the fact that Rh(II) is highly selective for
alkynes whose termini are not electronically differentiated (e.g. alkyl, see
row 2). Rh(II) can also tolerate alkyl-halides which
will be important for subsequent annulations etc. in the formation of more
elaborate natural product skeletons.
required forcing temperatures (>95 oC). Under these conditions only the stereo-electronically
favored 5-exo-dig cyclization was observed followed by isomerization. Thus we
set out to develop a more mild cyclization procedure
that would allow us to A) selectively generate the 5-exo-dig or
the 6-endo-dig product and B) maintain the fidelity of the alkene
position so that it can be used for further functionalization. Under the
simplistic assumption that a p-Lewis acid might
trigger the hydroamination process, a series of metal
salts were evaluated for their ability to affect the cyclization of an intact N,N-diboc-propargylguanidine
(Figure 6, Angew. Chem. Int. Ed.
2011, 50, 684-687). In
short we found that Ag(I) catalysts were optimum for
the formation of the formal 5-exo-dig product. We also discovered a
unique role of dirhodium carboxylates to promote the
selective formation of the formal 6-endo-dig product. Of particular
note is the fact that Rh(II) is highly selective for
alkynes whose termini are not electronically differentiated (e.g. alkyl, see
row 2). Rh(II) can also tolerate alkyl-halides which
will be important for subsequent annulations etc. in the formation of more
elaborate natural product skeletons.
