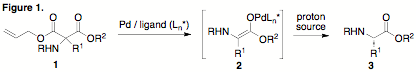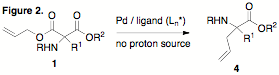AmericanChemicalSociety.com
Reports: DNI1 48922-DNI1: Asymmetric Decarboxylative Protonation of Alpha-Aminomalonic Esters for the Synthesis of Alpha-Amino Acids
a-Amino acids (a-AAs) are among the most important and useful families of natural products. The objective of our PRF-funded research project is to develop a novel synthetic method for the chemical synthesis of a-AAs that combines classical aminomalonate alkylation/decarboxylation with modern asymmetric catalysis. Specifically, we proposed the alkylation of unsymmetrical aminomalonic esters in which one of the ester substituents is an allyl group (1). The resulting compound 1 can undergo selective decarboxylation at the allyl ester using palladium catalysis to generate an intermediate enolate 2. We further proposed that the judicious selection of a chiral ligand for the palladium catalyst and an appropriate proton source would allow enantioselective protonation of the intermediate enolate giving rise to enantiomerically enriched a-AAs 3. This strategy is operationally straightforward with the potential for a more general substrate scope than existing methods.
Significant
progress has been made toward this overall objective during the first year of
this project. An efficient synthesis of the target aminomalonic esters 1 was developed and substrates were synthesized in
which R1 was H, CH3, and Bn. These substrates were
subjected to treatment with Pd under a number of different conditions. With R1
= H, it was observed that treatment with Pd and the (S)-tBu-PHOX ligand
resulted in rapid deallylation, but that efficient decarboxylation to provide
the desired amino acid 3 was
slow and required heating to 80 ¼C. When heated to this temperature in the
presence of Meldrum's acid as a proton source, the desired glycine ethyl ester
was isolated in 40–94% yield, depending on the solvent used (THF = 40%,
toluene = 38%, DMF = 65%, MeCN = 94%). When R1 = CH3, the
yields were reduced (DMF = 87%, MeCN = 73%); yields were further reduced when R1
= Bn (DMF = 45%, MeCN = 34%). These results imply that there is a possible
steric effect that influences the efficiency with which
decarboxylation/protonation occurs. Enantioselectivity of these reactions was
poor and we hypothesize that the slow rate of protonation relative to the very
rapid rate of deallylation renders the intermediate enolate decoupled from the
chiral Pd/ligand complex, thus resulting in a racemic protonation event. In addition to
these studies, we treated aminomalonic esters 1 with Pd in the absence of any proton source. It was observed that C-allylation
products 4 were obtained in good
yields. Quaternary amino acids of this type are also highly valuable materials
and thus this methodology represents a highly convenient strategy to provide
access. The presence of allyl functionality provides a versatile synthetic
handle for further synthetic modification. This
deallylation/decarboxylation/reallylation cycle is not enantioselective. It
also requires high temperature and it is likely that the deallylation and
decarboxylation occur in distinct steps, with Pd dissociated from the putative
enolate intermediate.
We reasoned
that substrates that undergo decarboxylation more readily would facilitate the
possibility of realizing this chemistry in an enantioselective fashion. One
method to improve decarboxylation propensity is to decrease electron deficiency
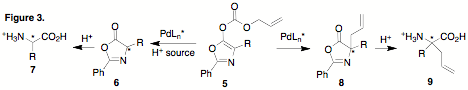
at the site of decarboxylation. In order to accomplish this we prepared cyclic
phenyloxazol-5-one derivatives 5. It
was reasoned that these derivatives have two advantages over the acyclic
malonates: 1) decarboxylation should be more facile; 2) the putative enolate
intermediate is cyclic, eliminating concerns with enolate geometry. These allyl
carbonates underwent rapid deallylation/decarboxylation and, in the absence of
a proton source, reallylation at the a-carbon
occurs in rapidly and in high yield (>90%), providing 8. These compounds can be readily hydrolyzed under
acidic conditions to give quaternary amino acids 9. We are in the process of determining the
enantioselectivity of these reactions and the second year of this funding will
be focused, in part, on optimizing enantioselectivity. We anticipate that this
will proceed initially by ligand screening in addition to perturbation of
reactions conditions.
When substrates
5 were treated with Pd in the presence
of Meldrum's acid as a proton source, very little of compound 6 was isolated. Instead, it was found that
reallylation to 8 was highly
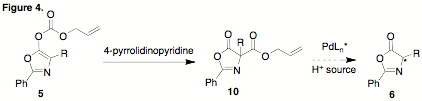
competitive with protonation. In the second year of this grant, we will attempt
to circumvent this competitive pathway. Our initial strategy will be to
rearrange 5 to 10. We will explore the reactivity of the malonate
relative to the carbonate in order to assess the possibility of efficient
protonation to form 6. Based on
our earlier observation that aminomalonates 1 undergo reasonably efficient protonation upon
deallylation/decarboxylation we wish to determine any possible differences in reactivity.
If reallylation proves to be highly efficient relative to protonation, other
substrate scaffolds will be explored in order to access amino acids.
Funding from
the Petroleum Research Fund has had a significant impact on my research
program. Our work is mainly focused on the study of peptide self-assembly
processes. We routinely use nonnatural amino acids as probes to perturb these
assembly processes and the development of novel and efficient methods to access
amino acids synthetically will be of great value to our overall research
efforts. The PRF has enabled us to undertake this work, which represents a new
research direction for our group and promises to make a major impact on our
program. The grant, to date, has supported the work of several students,
including summer research by an undergraduate student. The grant has
peripherally supported work that has resulted in three publications, and we
anticipate that the major objectives of the grant will be prepared for
publication in the next six months.