AmericanChemicalSociety.com
Reports: AC1 48511-AC1: Palladium(II)-Catalyzed Asymmetric Synthesis of alpha-Substituted Ketones
The
aim of this project is to develop new and more efficient methods of asymmetric
synthesis utilizing organopalladium catalysis, both for the enantioselective
introduction of new stereocenters alpha to a ketone carbonyl, and also for
intramolecular chirality transfer for a molecule with existing
stereocenters. Enantioselective
alpha-hydroxylation of ketones is traditionally accomplished through the
enantioselective oxidation of of enolate anions and enol derivatives such as
enol ethers or enol stannanes. We now
describe the direct enantioselective a-hydroxylation of ketones via the in-situ
generated enol tautomer using a bimetallic achiral Pd(II)
catalyst with triketone and chiral bidentate bridging groups with no need to form a base-generated
enolate or an enol ether. The
oxidation proceeds well with (R)-BINAP as the chiral ligand, and with 2,4,6-heptanetrione also present as a ligand.
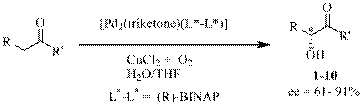
Run
| R
| R'
| % e.e.
| [alpha]20D
| |||||
1
| — CH2CH2CH2CH2—
| —
| 67
| 14.0
| |||||
2
| CH3
| Ph
| 68
| 62.3
| |||||
3
| CH3
| Ph
| 82
| 69.8
| |||||
4
| CH3
| Ph
| 51
| 46.2
| |||||
5
| CH3CH2
| Ph
| 71
| 23.7
| |||||
6
| Ph
| Ph
| 87
| 141.8
| |||||
7
| Ph
| Ph
| 85
| -139.9 [ligand = (S)-BINAP]
| |||||
8
| CF3
| Ph
| 89
| -7.8
| |||||
9
| 2-furyl
| 2-furyl
| 91
| 59.2
| |||||
10
| 3,5-difluorophenyl
| 3,5-difluoroPh
| 90
| 46.8
| |||||
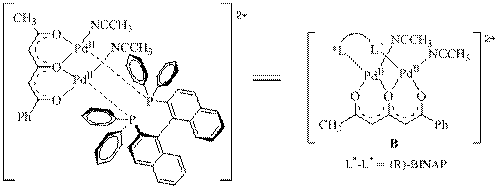
We believe the catalyst to be a bimetal structure as shown.
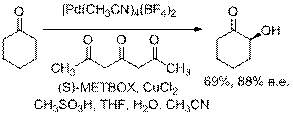
More recently, we have discovered that (S)-METBOX (2,2'methylenebis[(4S)-4-tert-butyl-2-oxazoline] provides even higher e.e.'s. In addition to cyclohexanone (90% yield, 88% e.e.), cyclopentanone, cycloheptanone, 2,5-dimethyl-4-heptanone, a-tetralone, propiophonenone and substituted propiophenone derivatives afford a-hydroxylated products in 84-94% e.e.

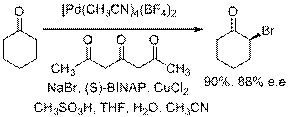
Asymmetric a-bromination has also been accomplished in high yield and very good e.e.'s using a bimetallic Pd(II) catalyst catalyst system.
We have extended this enantioselective a-functionalization reaction to the direct preparation of enantiomeric a-azido ketones using trimethylsilyl azide which will be extended to the synthesis of chiral vicinal amino alcohols.
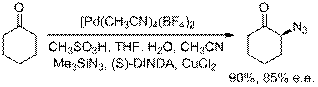
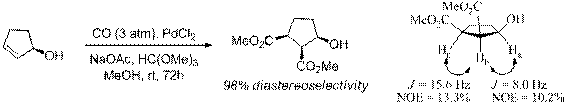
Finally, we have also explored chirality transfer from chiral allylic alcohols utilizing Stille's palladium-catalyzed olefin dicarbonylation reaction for the preparation of molecules with three contiguous chiral centers. (R)-(+)-2-Cyclopenten-1-ol afforded (1S,2S,3R)-dimethyl 3-hydroxycyclopentane-1,2-dicarboxylate in 65% yield and 98% diastereoselectivity. (R)-(Z)-3-Penten-2-ol gave (2R,3R,4R)-3,4-dicarbomethoxy-2-pentanol in 80% yield and 96% diastereoselectivity, whereas the geometric isomer (E)-3-penten-2-ol afforded (2R,3R,4S)-3,4-dicarbomethoxy-2-pentanol in lower yield (45%) and diastereoselectivity (78%).
Support for this project has enabled us to achieve proof of concept of the Pd(II)-mediated direct enantioselective a-hydroxylation reaction and to explore its generality, as well as to pursue the a-bromination and to discover the enantioselective a-azidation reactions. Separately, we have explored the Pd(II)-mediated diastereoselective bis-methoxycarboxylation reaction for the preparation of molecules with three contiguous chiral centers. This support by ACS-PRF has supported three post-doctoral scientists and enabled one student to make significant progress toward his Ph.D. degree.
