AmericanChemicalSociety.com
Reports: DNI1 49533-DNI1: Reaction Design for Nucleophilic Fluorination with Chiral Lewis Base Catalysts
Fluorine-containing organic molecules serve as critical pharmaceuticals, agrochemicals, performance materials, and radiotracers for imaging processes. Unfortunately, the availability of methods for the construction of C–F bonds severely limits the discovery and synthesis of these important targets. Particularly challenging in this context is the development of methods for the mild and selective incorporation of nucleophilic fluoride, the naturally occurring and least expensive form of fluorine, into organic targets. Use of anhydrous HF is plagued by difficulties with its safety and handling, metal fluorides suffer from sparing solubility in organic solvents, and reagents with enhanced solubility like tetraalkylammonium fluorides display high Brznsted basicity.
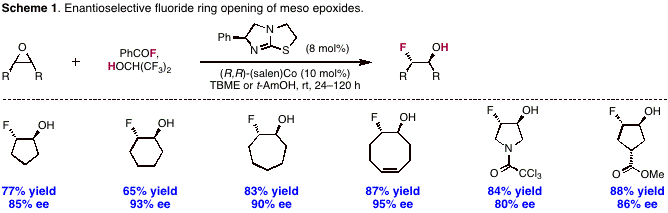
Instead, we hypothesized that commercial benzoyl fluoride could serve as bench-stable, organic-soluble latent fluoride source for catalytic C–F bond-forming reactions. In particular and in analogy to well-developed Lewis base-catalyzed acyl transfer, we proposed that a Lewis base catalyst could liberate HF from the combination of benzoyl fluoride and an alcohol additive. Because the uncatalyzed solvolysis of acyl fluorides is slow, we expected that (1) a catalytic procedure could overcome background reaction and (2) the protocol would be amenable to an enantioselective process through the intermediacy of chiral Lewis acid/amine co-catalysis. We first chose to evaluate this proposal in the context of epoxide fluorination, which provides access to synthetically versatile b-fluoro alcohols. In the event, we determined that commercial (–)-tetramisole and (R,R)-(salen)Co serve as co-catalysts for desymmetrizations of five- through eight-membered epoxides, affording b-fluoro alcohols in up to 95% ee (Scheme 1).
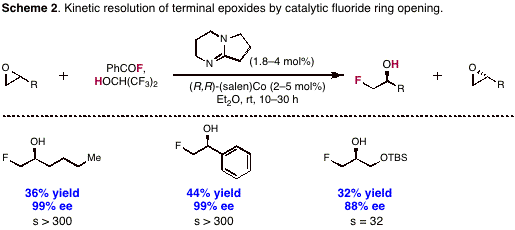
We have also explored terminal
epoxides as substrates for fluoride ring-opening kinetic resolutions and found
that a broad range of substrates undergo reaction with extremely high
regioselectivity and krel
values (Scheme 2). In these reactions, 1,5-diazabicyclo[4.3.0]non-5-ene (DBN)
was identified as the optimal amine co-catalyst. Notably, the co-catalytic
fluorinations proceed at room temperature in standard glassware and do not
require exclusion of moisture or air. A consequence of these mild conditions is
that substrates containing traditionally fluoride-labile substituents such as
silyl ethers are stable to the reaction conditions and undergo C–F bond
formation with good reaction efficiency.
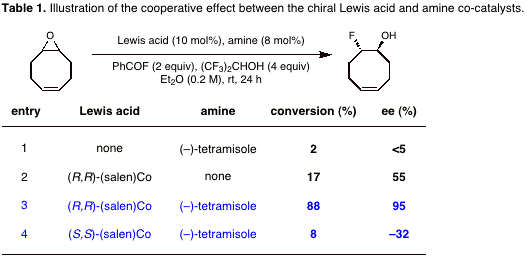
The fluorinations with
(–)-tetramisole and (salen)Co(II) in Scheme 1 exhibit a pronounced
matched/mis-matched effect on rate and enantioselectivity (Table 1). Although
amine/Lewis acid co-catalysis has been described, a cooperative effect on
enantioselectivity has little precedent. We are currently undertaking kinetic
and non-linear effect studies in order to elucidate the role of the two
catalysts in the catalytic fluorinations. We anticipate that these studies will
lead to improvements to the catalytic methodology and to the development of new
methodologies for the incorporation of fluorine into organic substrates. Along
these lines, we have begun to undertake studies aimed at the development of an
enantioselective fluoride ring-opening reaction of aziridines. The Doctoral New
Investigator grant from the ACS PRF has been instrumental in jump-starting this
program and we are extremely grateful for the support.