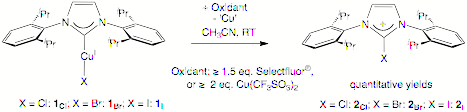AmericanChemicalSociety.com
Reports: AC5 48526-AC5: Click-On SBA-15: Efficient Variable Loading of Oxidation Catalysts in Porous Silica
N-heterocyclic carbene (NHC) metal complexes are used widely as catalysts in organic reactions. Compared to other neutral type ligands, NHCs usually form stronger bonds with metals due to excellent s donating ability. The conjugation between the carbene carbon (Ccarbene) pp orbital and nitrogen lone pair electrons in the heterocycle further stabilizes the metal–Ccarbene bonds. As a result, NHCs are often better at suppressing catalyst decompositions. The metal–Ccarbene bonds are remarkably inert compared to other metal–carbon bonds at catalytic metal centers, which are prone to undergo various reactions such as migratory insertion and olefin metathesis. In contrast, only a small number of elementary organomellic reactions, mostly limited to Ccarbene–C reductive elimination and ligand dissociation/displacement, are documented with the metal–Ccarbene bonds.
Carbon-halogen (C–X) reductive elimination of metal–Calkyl or metal–Caryl bonds is an important elementary organometallic reaction, but no well-defined example is reported for Ccarbene–X reductive elimination from NHC metal halide complexes, although such complexes are ubiquitous in NHC chemistry. Herein, we report facile formation of 2-halo-imidazoliums from NHC Cu(I) halide complexes at room temperature (RT) under oxidative conditions as well as computational evidence to support a mechanism involving Ccarbene–X reductive elimination from NHC Cu(III) halide complexes.
As a part of our
efforts in the characterization of reactivity of copper(III) complexes, we
postulated that NHC Cu(III) complexes might be isolable because of the
relatively inert Cu–Ccarbene bond. In that regard, oxidations
of IPrCuICl (1Cl, IPr: 1, 3-bis(2,
6-diisopropylphenyl)imidazol-2-ylidene) by various oxidants were investigated.
No significant reaction was observed between 1Cl and [Ph2I]+PF6-
or [Cp2Fe]+PF6- (0.64 V vs NHE) in acetonitrile (MeCN). By
contrast, mixing 1Cl with Selectfluor¨ (³ 1.5 eq.) or CuII(CF3SO3)2
(³ 2.0 eq., 1.30 V vs NHE) in MeCN at RT rapidly and quantitatively forms
2-chloro-imidazolium 2Cl (Scheme 1). Quantitative formation of 2-halo-imidazolium 2Br or 2I was realized for IPrCuIBr
(1Br)
or IPrCuII (1I) under similar conditions. These reactions appear to occur through either
an inner- or outer-sphere oxidation followed by Ccarbene-X reductive
elimination. An outer-sphere oxidation is supported by the ca. 80 % formation of 2Cl by reacting
1Cl with [(1, 10-phenanthroline)3FeIII]3+
(³ 2 eq., 1.22 V vs NHE) in MeCN at RT. However, all these reactions are fast even at low
temperatures (t1/2 ~ seconds at ca. -40 °C), and no intermediate species
could be detected by UV-Vis spectroscopy on a timescale of seconds. DFT calculations
provide mechanistic insights into the oxidation by Selectfluor¨. The
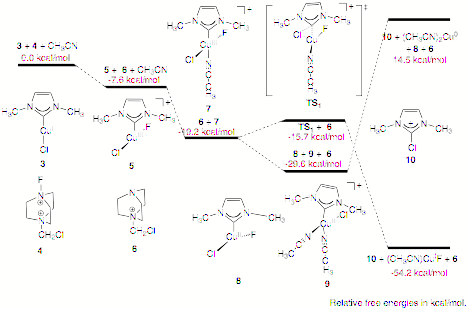
calculated free-energy profiles corresponding to Ccarbene-X
reductive elimination from either a Cu(II) or Cu(III) species using a
simplified model system (3) are shown in Scheme 2. The oxidation of 3 by Selectfluor¨ to form 5, a three-coordinate Cu(III)
species, is thermodynamically favorable. The Cu(III) species is further
stabilized by coordination of an MeCN ligand to form 7. The activation barrier for Ccarbene–Cl
reductive elimination from 7 via TS1 to form 2-chloro-imidazolium 10 is remarkably low at 3.5 kcal mol-1.
The overall reaction is favorable by -54.2 kcal mol-1. Other
possible Cu(III) species are less stable than 7 and lead to higher activation
barriers. Alternatively, 7 could react with another equivalent of 3 to form Cu(II) intermediates 8 and 9, the two most stable Cu(II) species. Although
no transition state from 9 to 10 could be located (the energy monotonically increases as the
Ccarbene–Cl separation shortens.), the lower limit of its
activation barrier can be estimated by the corresponding reaction free energy,
44.1 kcal mol-1, the least endothermic among various Cu(II) species.
Therefore, the calculations on the simplified model system favor a mechanism of
Ccarbene–Cl reductive elimination from NHC Cu(III) chloride
complexes for the oxidation of 1Cl by Selectfluor¨.
The thermodynamically unfavorable Ccarbene–Cl
reductive elimination from 9 is consistent with isolable NHC Cu(II) chloride complexes
reported in the literature. Based on these results, we prefer a mechanism with
two sequential inner- or outer-sphere 1e- oxidations followed by Ccarbene–Cl
reductive elimination from NHC Cu(III) chloride complexes for the reaction with
Cu(CF3SO3)2, although the detailed mechanism
is still unclear. Furthermore, the quantitative formation of 2Cl instead of IPrCu(II) chloride
complexes from the reaction of 1Cl and Selectfluor¨
suggests that Ccarbene–Cl reductive elimination from
IPrCu(III) chloride complexes is much faster than reactions of 1Cl to form IPrCu(II) halide complexes.
This reactivity is consistent with the calculated 3.5 kcal mol-1
activation barrier from 7 to 10. Such a low barrier is probably a consequence of the
electrophilic nature of a Cu(III) center that renders the NHC Ccarbene
susceptible to nucleophilic attack, as suggested by the remarkably close Ccarbene–Cl
contacts (ca.
2.7) in 5 and 7 as well as the interactions between
the chloride lone pair electrons and the Ccarbene pp orbital in relevant molecular
orbitals. A similar interaction has been invoked to rationalize the short Ccarbene–Cl
contact (ca.
2.85) in NHC VV(O)Cl3. In contrast, no evidence of such
an interaction exists for 8. In summary, we have demonstrated that Ccarbene–halogen
reductive eliminations readily occur from NHC copper halides at RT under
oxidative conditions. These reactions provide new examples for the well-known
oxidation-induced reductive eliminations. DFT calculations on a simplified
model system suggest that the involvement of NHC Cu(III) halides is essential
for these reactions and the reductive eliminations might be facilitated by the
interaction between Ccarbene and the halogen lone pair. Given the
ubiquity of NHC metal halide complexes, the facile Ccarbene–X
reductive elimination reported here warrants consideration as a potential
decomposition pathway in reactions involving NHC-supported high-valent metal
complexes, especially with late transition metals.