AmericanChemicalSociety.com
Reports: AC1 47306-AC1: Diels-Alder Cycloaddition Reactions with Dihapto-Coordinated Pyridines
Part I. Cycloaddition Reactions
The constructions of numerous polyheterocyclic scaffolds has been achieved utilizing hetero Diels-Alder reactions. In particular, the isoquinuclidine skeleton, found in a variety of alkaloids (e.g., ibogaine, 1) and their pharmaceutically-derived analogs, has been prepared using aza-butadienes, dihydropyridines, or pyridones, combined with various dienophiles. We reasoned that common pyridines would be attractive alternatives as sources of azadienes, but their aromatic stability normally renders them unsuitable for such cycloaddition reactions. Our research group has been exploring the effects of dihapto-coordination on the chemistry of aromatic molecules. Once coordinated, the aromatic ring is dearomatized, activating the aromatic toward new chemical transformations. We postulated that if 2,6-dimethoxypyridine (h2-DMP) were bound to {TpW(NO)(PMe3)}, the uncoordinated portion of this ring should take on a chemical nature similar to that of Danishefsky's diene. Specifically, the combination of ¹-donation from both tungsten and the methoxy groups was expected to render this pyridine a potent reagent for cycloaddition reactions. The use of a dihapto-coordinated 2,6-dimethoxypyridine allows access to a full range of alkene and alkyne dienophiles, and in some cases, these azabicyclic skeletons can be chemically modified while bound to the tungsten fragment prior to their decomplexation. The results of this study have now been now been published: Organometallics, 2008, 27, pp 4513–4522.
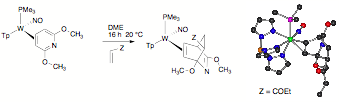 Figure
1.
Figure
1.
Part II. The Synthesis of Highly Functionalized Piperidines from Pyridines
During the investigation of Diels-Alder reactions with dihapto-coordinated pyridines, we developed a method for forming the parent pyridine complex (trapped as its conjugate acid) from pyridine borane. While the parent dihapto-coordinate pyridine complex failed to undergo Diels-Alder reactions fast enough to overcome the isomerization to the N-bound isomer, isolation of this complex was a real breakthrough as it allowed a route to other pyridinium complexes such as the N-acetylated pyridinium complex (Figure 1).
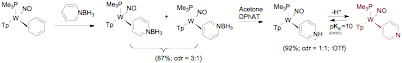 Figure 2.
Figure 2.
With unsubstituted pyridinium complexes finally in hand, the focus shifted to uncovering new reaction sequences for tungsten-pyridine complexes (Path 1 and 2, Figure 2).
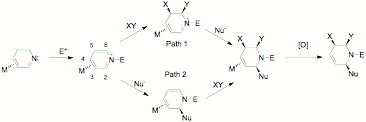 Figure 3.
Figure 3.
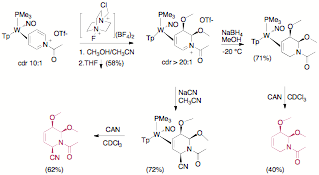
Path 1. Dihapto-coordinated pyridinium complexes of {TpW(NO)(PMe3)} (E = Ac) were found to undergo a stereoselective dialkoxylation reaction when treated with Selectfluor¨ in an alcohol. The alkoxy groups add to the 5 and 6 positions in a syn fashion (proven by X-ray diffraction). When the N-acetylpyridinium complex is dialkoxylated, the resulting acyldihydropyridinium system readily undergoes nucleophilic addition with cyanide ion to stereoselectively generate a third stereocenter at C2 of the pyridine ring. Treatment with CAN decomplexes the tetrahydropyridine (Figure 4). These findings have been published: Organometallics, 2009, 28, 387–389.
 Figure
4.
Figure
4.
Path 2. The complimentary reaction sequence was next explored: nucleophilic addition at C2 followed by elaboration at C5 and C6. The N-acetylated pyridinium complex readily undergoes regio- and stereoselective nucleophilic addition, exclusively at C2, and anti to the tungsten. This reaction occurs with a remarkably broad range of nucleophiles including enolates, acetylides, alkyllithiums, alkylzincs, hydrides, cyanide, and nitrogenous heterocycles (indole, pyrrole, etc; Figure 5). These results have been published: Organometallics, 2009, 28, pp 5682–5690.
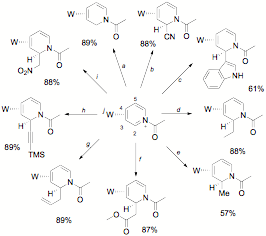 Figure 5.
Figure 5.
With access to a broad range of 2-substituted h2-1,2-dihydropyridine complexes, we endeavored to functionalize the remaining double bond at C5-C6. Enamides, like enamines, polarize the alkene bond such that electrophiles normally add to the b carbon. However, studies of h2-coordinated 1,3-dienes show that electrophilic addition typically occurs at the uncoordinated terminal alkene carbon. Thus, 1,2-dihydropyridine complexes with the metal at C3,C4 present an interesting regiochemical question, pitting the electron donation of the amide nitrogen against that of the ¹ basic tungsten complex. Here we find that protonation occurs exclusively at C6 (an umpolung), resulting in a ¹ allyl complex.
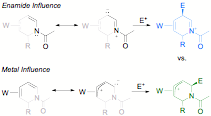 Figure
6.
Figure
6.
This allyl species was found to be highly asymmetric, both in terms of its electronic character and its chemical structure. The bond lengths of tungsten to the two terminal allyl carbons differ by almost 0.4, with C3 taking on considerable free carbocation character. Correspondingly, C3 of the pyridine ring is readily attacked by a range of nucleophiles generating 5,6-disubstituted D3-piperidine complexes. In certain cases, steric factors direct the second nucleophilic addition to C5 of the pyridine ring, resulting in 2,5-disubstituted D3-piperidines. These observations of nucleophilic addition to C3 or C5 of a pyridine ring system represent an umpolung of the natural chemical behavior of this heterocycle, allowing for the preparation of unique substitution patterns not easily available by conventional methods. Six of these compounds are currently being evaluated for medicinal use (Johns Hopkins, Broad Institute, Scripps). A manuscript describing the details of this process has been submitted for publication.
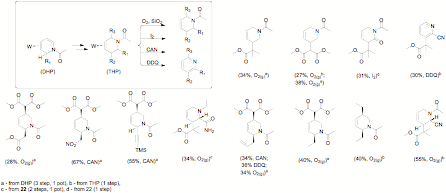 Figure 7.
Figure 7.
Part III. Ring-opening of the pyridine core.
Cyanines and merocyanines, polyene systems with an electron donor group at one terminus and a withdrawing group at the other, are widely studied not only in the historical context of the dye industry, but for their non linear optical and solvatochromism properties, and their potential uses in laser technology, data storage, photosensitizers, and phototheraputics. In the course of investigating the reactivity of h2-coordinated pyridinium complexes of tungsten, we came across an unexpected reaction pathway in which the bound pyridinium ligand, upon treatment with certain nucleophiles, spontaneously underwent ring scission, cleanly forming a new type of "metallocyanine" (Figure 8). These results were recently published: Organometallics, 2010, 29, 1909.
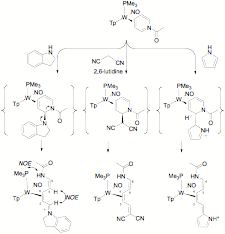 Figure
8.
Figure
8.
Part IV. Novel Dihydropyridine Cycloadditions.
The dihydropyridine complexes described in Figure 5 were found to be attractive precursors to novel bicyclic alkaloids. While 1,2-dihydropyridines would normally undergo cycloaddition in a manner consistent with the C3 and C6 carbons being polarized negative and positive respectively, metal coordination reverses this polarization. Preliminary results are shown in Figure 9.
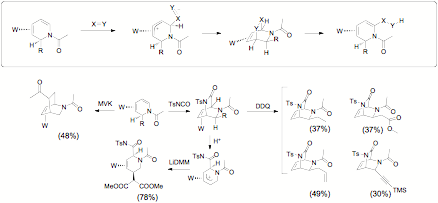 Figure 9.
Figure 9.
