

45685-G3
An Electrochemically Controlled Separation of Nitrogen and Sulfur Heterocycles from Crude Petroleum by Reversible Binding to Iron Bis(dithiolene) Complexes
The funded research was initiated as an effort to explore redox-controlled, reversible ligand binding to metallodithiolene complexes as a possible means to separate sulfur and nitrogen heterocyclic molecules from crude petroleum streams. Such molecule types, if left unremoved and burned, contribute to pollution. Their potential to act as ligands provides an alternative to the hydrotreating process, which is energy intensive and can alter the octane rating of refined fuel.
Another route
to sterically hindered dithiolene ligands that was investigated was that
proceeding through 4,5-dialkyl-1,3-dithiol-2-one, R2C2S2C=O.
In the course of this study, a simplified and improved synthesis of Me2C2S2C=O
was made, which enabled its facile preparation on a scale of tens of grams. The
synthesis of [Ni(S2C2Me2)2] was
tested using 4,5-dimethyl-1,3-dithiol-2-one, Me2C2S2C=O
and found to be far cleaner and higher yielding (87% yield) than the original P4S10/acetoin
method of Schrauzer. Conversion of Me2C2S2C=O to
(Me2C2S2)SnnBu2,
followed by reaction with WCl6 produced tris(dithiolene) [W(S2C2Me2)3]
in 80% yield, a similar improvement over the P4S10/acetoin
method and demonstrated the potential synthetic utility of this tin-protected
form of the ligand in metallodithiolene synthesis. The series of compounds [W(S2C2Me2)x(CO)6-2x]
(x = 1-3), obtained as a mixture via the reaction of [Ni(S2C2Me2)2]
with [W(MeCN)3(CO)3], was characterized structurally. The
structure of [W(S2C2Me2)(CO)4] (4) was observed to be trigonal
prismatic, a geometry confirmed by DFT to be lower in energy than an octahedron
by 5.1 kcal/mol. The reactivity of 4
and [W(S2C2Me2)2(CO)2] (5) toward PMe3 was examined.
Compound 4 yielded [W(S2C2Me2)(CO)2(PMe3)2],
while 5 produced either [W(S2C2Me2)2(CO)(PMe3)]
(8) or [W(S2C2Me2)2(PMe3)2]
(9) depending upon conditions. Characterization
by X-ray crystallography of 5, 8, and 9 revealed a trend toward greater reduction of the dithiolene
ligand across the series, as manifested by C-C and C-S bond
lengths. These structural data indicated a profound effect exerted by the
π-acidic CO ligands upon the apparent redox state of the dithiolene ligand.
 For
ease and convenience, initial work made use of phosphine ligands rather than
sulfur or nitrogen heterocycles. The homoleptic bis(dithiolene) complexes [M(S2C2R2)2]2
(M = Fe, Co; R = p-anisyl) were
studied and found to undergo two successive reductions to form anions that
display [M(S2C2R2)2]22-
↔ 2[M(S2C2R2)2]1-
solution equilibria. The neutral dimers reacted with Ph3P to form
square pyramidal [M(Ph3P)(S2C2R2)2]0.
Voltammetric measurements upon [M(Ph3P)(S2C2R2)2]0
in CH2Cl2 revealed only irreversible features at negative
potentials, consistent with Ph3P dissociation upon reduction.
Dissociation and re-association of Ph3P from and to [Fe(Ph3P)(S2C2R2)2]n
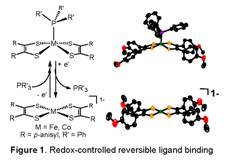
was demonstrated by spectroelectrochemical measurements (Figure 1). These
collective observations form the basis for a cycle of reversible, electrochemically
controlled binding of Ph3P to [M(S2C2R2)2]2
(M = Fe, Co; R = p-anisyl). All
members of the cycle ([M(S2C2R2)2]20,
[M(S2C2R2)2]21-,
[M(S2C2R2)2]22-,
[M(S2C2R2)2]1-, [M(Ph3P)(S2C2R2)2])
for M = Fe, Co were characterized by crystallography. Square planar [Fe(S2C2R2)2]1-
was the first such iron dithiolene species to be structurally identified. Thermochemical
aspects of this redox-controlled ligand binding were considered with DFT
calculations.
For
ease and convenience, initial work made use of phosphine ligands rather than
sulfur or nitrogen heterocycles. The homoleptic bis(dithiolene) complexes [M(S2C2R2)2]2
(M = Fe, Co; R = p-anisyl) were
studied and found to undergo two successive reductions to form anions that
display [M(S2C2R2)2]22-
↔ 2[M(S2C2R2)2]1-
solution equilibria. The neutral dimers reacted with Ph3P to form
square pyramidal [M(Ph3P)(S2C2R2)2]0.
Voltammetric measurements upon [M(Ph3P)(S2C2R2)2]0
in CH2Cl2 revealed only irreversible features at negative
potentials, consistent with Ph3P dissociation upon reduction.
Dissociation and re-association of Ph3P from and to [Fe(Ph3P)(S2C2R2)2]n
was demonstrated by spectroelectrochemical measurements (Figure 1). These
collective observations form the basis for a cycle of reversible, electrochemically
controlled binding of Ph3P to [M(S2C2R2)2]2
(M = Fe, Co; R = p-anisyl). All
members of the cycle ([M(S2C2R2)2]20,
[M(S2C2R2)2]21-,
[M(S2C2R2)2]22-,
[M(S2C2R2)2]1-, [M(Ph3P)(S2C2R2)2])
for M = Fe, Co were characterized by crystallography. Square planar [Fe(S2C2R2)2]1-
was the first such iron dithiolene species to be structurally identified. Thermochemical
aspects of this redox-controlled ligand binding were considered with DFT
calculations.
 Observations
made in the foregoing studies suggested that weakly-coordinating ligands such
as thiophenes would be unable to disrupt the [M(S2C2R2)2]2
dimer and bind as an axial ligand. This problem led to a consideration of how
the dithiolene ligand substituents might be modified in a way to disfavor
dimerization but still permit a weak axial ligand interaction. Ideal dithiolene
ligand substituents would be sterically hindered and electron deficient. The
lack of synthetic routes to such ligands led to a study of ways by which
dithiolene ligands, or ligand precursors, might be more readily prepared. It
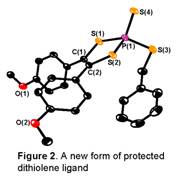
was found that reaction of P4S10 with acyloins,
RC(O)CN(OH)R, in refluxing dioxane, followed by the addition of alkylating
agents, forms dithiolene thiophosphoryl thiolate compounds, (R2C2S2)P(=S)(SR
Observations
made in the foregoing studies suggested that weakly-coordinating ligands such
as thiophenes would be unable to disrupt the [M(S2C2R2)2]2
dimer and bind as an axial ligand. This problem led to a consideration of how
the dithiolene ligand substituents might be modified in a way to disfavor
dimerization but still permit a weak axial ligand interaction. Ideal dithiolene
ligand substituents would be sterically hindered and electron deficient. The
lack of synthetic routes to such ligands led to a study of ways by which
dithiolene ligands, or ligand precursors, might be more readily prepared. It
was found that reaction of P4S10 with acyloins,
RC(O)CN(OH)R, in refluxing dioxane, followed by the addition of alkylating
agents, forms dithiolene thiophosphoryl thiolate compounds, (R2C2S2)P(=S)(SR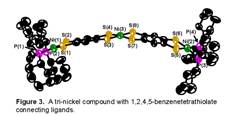 Finally,
in related work, the PI engaged in some exploratory synthesis of complex,
multimetal dithiolene complexes. The controlled base hydrolysis of one or both
of the protected 1,2-dithiolene chelates of 1,3,5,7-tetrathia-s-indacene-2,6-dione (O=CS2C6H2S2C=O) enabled the stepwise
synthesis of di- and trimetallic complexes with 1,2,4,5-benzenetetrathiolate as
connector. Treatment of O=CS2C6H2S2C=O
with MeO-, followed by
[NiBr2(dcpe)] (dcpe =
1,2-bis(dicyclohexylphos-phino)ethane) yielded [(dcpe)Ni(S2C6H2S2C=O)] (4). Reaction of 4 with
EtO- followed by [MX2(dcpe)] produced [(dcpe)Ni(S2C6H2S2)M(dcpe)] (M = Ni (5a), Pd (5b)). Deprotection of the 1,3-dithiol-2-one group of 4, followed by introduction of ˝ eq of
MX2 and then I2,
yielded the neutral trimetallic compounds [(dcpe)Ni(S2C6H2S2)]2M (M = Ni (6a), Pt (6b)), Figure 3. A color darkening was observed in moving from 4 to compounds 6 due to the increasing size of the conjugated metal-organic π-system.
Intense absorptions in the near IR were observed for compounds 6ab.
Finally,
in related work, the PI engaged in some exploratory synthesis of complex,
multimetal dithiolene complexes. The controlled base hydrolysis of one or both
of the protected 1,2-dithiolene chelates of 1,3,5,7-tetrathia-s-indacene-2,6-dione (O=CS2C6H2S2C=O) enabled the stepwise
synthesis of di- and trimetallic complexes with 1,2,4,5-benzenetetrathiolate as
connector. Treatment of O=CS2C6H2S2C=O
with MeO-, followed by
[NiBr2(dcpe)] (dcpe =
1,2-bis(dicyclohexylphos-phino)ethane) yielded [(dcpe)Ni(S2C6H2S2C=O)] (4). Reaction of 4 with
EtO- followed by [MX2(dcpe)] produced [(dcpe)Ni(S2C6H2S2)M(dcpe)] (M = Ni (5a), Pd (5b)). Deprotection of the 1,3-dithiol-2-one group of 4, followed by introduction of ˝ eq of
MX2 and then I2,
yielded the neutral trimetallic compounds [(dcpe)Ni(S2C6H2S2)]2M (M = Ni (6a), Pt (6b)), Figure 3. A color darkening was observed in moving from 4 to compounds 6 due to the increasing size of the conjugated metal-organic π-system.
Intense absorptions in the near IR were observed for compounds 6ab.