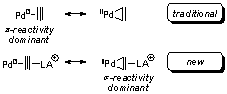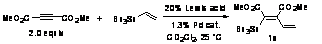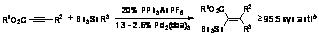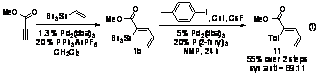46285-G1
Nucleophilic Substitution/Functionalization Reactions of Transition-Metal Complexes at sp3 Carbon
Palladium-catalyzed carbon-carbon cross-coupling reactions are a staple of modern synthetic chemistry due to the diverse products accessible and functional groups tolerated by these reactions. While powerful, these methods require initial oxidative addition into a carbon-halogen, -oxygen, or -hydrogen covalent (or ionic) bond (e.g., Stille, Heck, Suzuki, Trost-Tsuji, Buchwald-Hartwig,1 and the ionic Arndtsen modification2). During the last ACS-PRF granting year, we reported the extension of this chemistry from C-X bonds to C-C p-bonds through the employment of alkynes in the place of halides in a Stille-type vinylstannylation reaction. Our reaction provides an expedient route to the monoaddition products, yielding tri- and tetra-substituted olefins3,4 from alkynes with absolute regioselectivity and high stereoselectivity via a Au(I)/Pd(0) bimetallic catalyst system that combines the characteristics of two metals to produce unique reactivity.5
We hypothesized that the alkynophilic6 Lewis acid Au(I) would lower the LUMO of an alkyne fragment, promoting backbonding (i.e., oxidative addition) from Pd(0) into the alkyne,7 which in turn increases palladium-carbon s-bond character (Figure 1). Andersen reported a conceptually similar, structurally characterized m-ethylene bimetallic complex with Pt(0) as the Lewis base and Yb(II) as the Lewis acid.7 To our knowledge, no employment of this structure class as a catalytic intermediate had been developed, despite the potential generality of such a method for accessing late-metal-carbon s-bonds. With an alkyne as the oxidative addition partner, no C-X bond would be necessary for a cross-coupling reaction (i.e., the alkyne serves as a pseudohalide).8
In order to explore this
hypothesis, dimethyl acetylenedicarboxylate (DMAD) was treated with 20 mol% of
PPh3AuCl/AgSbF6 and 5 mol% Pd2(dba)3
in the presence of one equivalent of tri-n-butylvinylstannane. Gratifyingly,
these conditions produced tetrasubstituted olefin 1a in 51% 1H
NMR yield. Both Au(I) and Pd(0) were required for conversion. Supporting Pd(0)'s
role as an oxidative addition partner to the alkyne, Pd(II) is significantly
less active.
The utility of the Pd/Au
catalyzed method for formation of tri-and tetra-substituted olefins is illustrated
by a range of stannane partners (i.e., sp2, sp,
secondary, oxygenated, b-substituted; Table 1). Complete regioselectivity is
maintained even in the presence of a sterically bulky t-butyl ester. Interestingly, no evidence for reentry into the catalytic
cycle by the product vinyl stannanes is observed. Presumably, this is because
of the increased steric hinderance provided by a branching and b substitution. A proposed mechanism for the
Au- and Pd-cocatalyzed vinylstannylation of alkynes is detailed in Scheme 1.
Coordination of cationic Au(I) to the alkyne promotes nucleophilic
addition/oxidative addition of Pd(0). Transmetallation of tri-n-butylvinylstannane
across one of the palladium-carbon s-bonds of 7
results in vinyl transfer to palladium and tin transfer to the nascent olefin.
Dissociation of Au(I) followed by reductive elimination forms the observed
vinylstannylated product, 10, and regenerates Pd(0).
We next investigated the
ability of the stannyldiene products to participate in traditional cross-coupling
reactions with aryl halides. Reaction of crude stannyldiene 1b with iodotoluene
in the presence of 5% additional Pd2(dba)3 yields the
all-carbon trisubstituted olefin 11 in 78% yield (55% over two steps)
(eq 1). This two-step procedure affects an aryl vinylation of the starting
alkyne with absolute regioselectivity and high stereoselectivity for the syn
addition product.
In conclusion, we have developed a palladium and gold
cocatalyzed vinylstannylation of alkynes to form tri- and tetra-substituted
olefins with excellent regio- and stereocontrol. The reactions are postulated
to proceed via Au(I) activation of the alkynes toward nucleophilic metallation/oxidative
addition to Pd(0). In this mechanism, the alkyne serves as a pseudohalide in a
Stille-type cross-coupling reaction. In a broader sense, the reactions
reported herein provide an entry into the extensive catalytic chemistry of
palladium-carbon s-bonds starting from p-systems. 1. Hartwig,
J. F. Acc. Chem. Res. 1998, 31, 852. 2. Davis,
J. L.; Dhawan, R.; Arndtsen, B. A. Angew. Chem. Int. Ed. 2004, 43,
590. 3. Itami,
K.; Kamei, T.; Yoshida, J. J. Am.