

44063-G3
Bridging the Gap Between Early and Late-Metal Catalysis: d2 Square-Planar Complexes That Mimic Their d8 Counterparts
`The intent of this proposal was to design new trianionic pincer ligands that support a square–planar geometry for group VI, d2 metals and elucidate the chemistry that this imposed geometry confers (Figure 1).
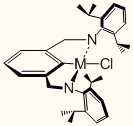
Figure SEQ Figure \* ARABIC 1. New Trianionic pincer ligand (iPrNCN3–)
bound to M, where M = W(IV) (1) and Mo(IV) (2).
As the proposed work progressed, it became obvious the square planar complex (Figure 1) would not be isolated with the NCN ligand. Eventually we synthesized the OCO trianionic pincer ligand depicted in figure 2.
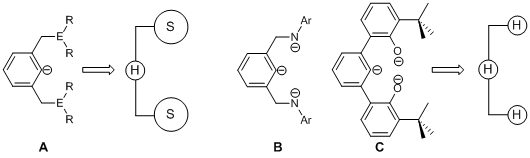
Figure 2. Monoanionic pincer ligand (A) versus trianionic pincer ligands (B and C).
Classic pincer ligands (A, E = P, and N) are complementary to late transition metals due to their soft–hard–soft arrangement of donor atoms. Our approach is to match the harder early transition metals with a harder pincer ligand. Instead of a soft–hard–soft pincer motif, we have synthesized a series of new hard–hard–hard pincer ligands based on amido–arylide–amido (B, Ar = 2,6–iPrC6H3, and 3,5–MeC6H3) and alkoxide–arylide–alkoxide linkages (C, OCO3– = 1,3-C6H4(6-tBuC6H3OH)2).
Ligand Attributes
- occupy three coordination sites but contribute maximum of 10e- (access electronically unsaturated species)
- rigid backbone allows only meridional coordination (access constrained, high- energy species)
- tridentate trianionic versus three individual monodentate monoanionic ligands (increased stability, resistant to protonation)
- easily adjust electronics, sterics, rigidity, and chelate ring size
New Chemistry
Restricting three anionic donor ligands to meridional positions generates reactive metal fragments. As proof, an [OCO]Mo-nitrido complex readily adds mild electrophiles and completes a N-atom transfer to acid chlorides to synthesize nitriles. The Hard-Hard-Hard donor combination allows access to unusual high oxidation states of Cr, namely Cr(IV) and Cr(V). Finally, preliminary evidence suggests reversible water addition across a [OCHO]W≡W[OCHO] triple bond leads to a rare N-N coupling reaction and a W-W quadruple bonded complex.
When a yellow-orange THF solution of 1 is treated with one equivalent of 2,6-lutidinium·HCl to 1, deep purple nitrido-amine [tBuOCO]Mo≡N(NHMe2)
(2) forms within 15 min at 23 °C (eq. 1). Analytically pure nitrido-amine 2 precipitates in 36% yield by dropping
a concentrated THF solution of the reaction mixture into cold pentane. The 1H NMR spectrum of 2 indicates the two diastereotopic
amido-methyl resonances of 1 (4.10
and 2.60 ppm) collapsed to a doublet at 2.36 ppm (J = 6 Hz), which integrates as the six amine protons.
The imposed
strain by the ligand and anionic charge elevates the reactivity of the terminal
nitride, thereby permitting the addition of electrophiles. Treating 1 with the mild electrophiles Me3SiCl
and MeI forms the silylimido [tBuOCO]Mo=NSiMe3(NMe2)
(3-SiMe3) and methylimido
[tBuOCO]Mo=NMe(NMe2) (3-Me)
complexes, respectively (eq. 2).
Ultimately we were able to show that the
N-atom could be completely liberated and transferred to acid chlorides to
synthesize various nitriles according to scheme 1. One of the stated goals of the proposal was
to achieve the difficult feat of N-atom transfer. Though we are still working on isolating a d2
square-planar complex the N-atom transfer portion of the proposal was
achieved. Moreover, we have new insight
into possible routes to activating dinitrogen. Water
addition across M-M multiple bonds. Water oxidation and catalysis will
be examined using new homogenous mono- and dinuclear complexes supported by the
OCO pincer ligand [tBuOCO]H3. This
ligand supports unusually reactive metal complexes and recently the Veige group
demonstrated water addition across a metal-metal triple bond (Eq 2.).
Provided below eq 2 is the structure of the resulting W-O-W complex
after H2O addition. The
reaction is unusual because the reducing equivalents come from the triple bond. As the reaction proceeds, a red precipitate
forms. A 1H NMR spectrum of
the supernatant revealed the formation of teramethylhydrazine, an N-N coupling
product and one equivalent of H2O.
This three-step cascade reaction – double O-H addition of water across a
W-W triple bond – N-N coupling to form Me2N-NMe2, and
double O-H reductive elimination is unprecedented. The proposed product is a W-W quadruple
bonded complex, though its identity and the reaction sequence has yet to be
fully elucidated.
The trianionic
pincer ligand also allows access to unusual, high oxidation state Cr species. These species are exceptionally rare and
their reaction chemistry is essentially unknown. The
Veige group now possesses complexes with oxidation states that range from
Cr(III)-Cr(IV)-Cr(V).
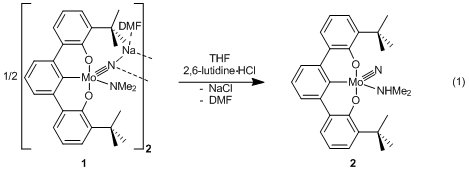
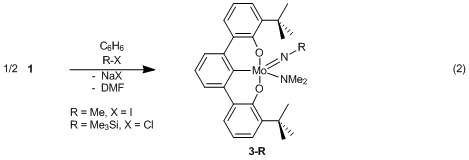
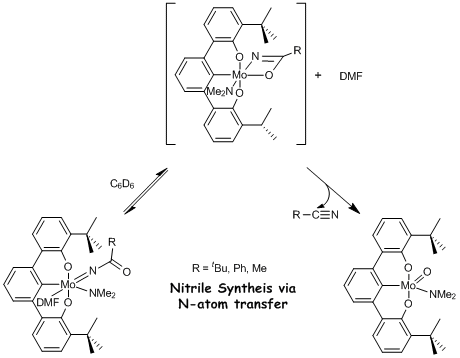
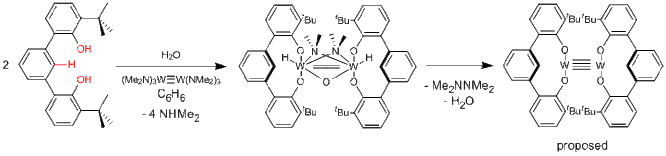
Figure 3. Single crystal X-ray structure of 3.
Figure 4. Single crystal X-ray structure of 4. 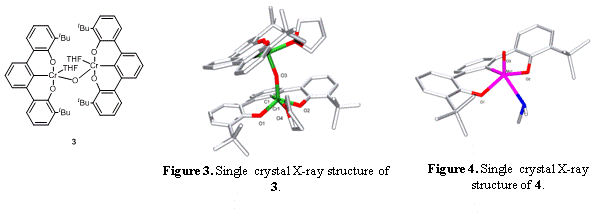
Figure 5.
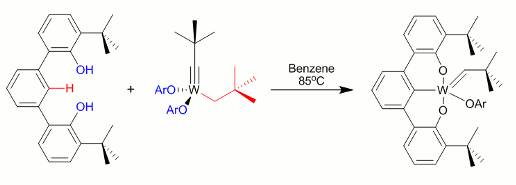
A final aspect of the explored chemistry involves M-C multiple bonds. We sought a four coordinate alkylidyne (see target complex above) supported by our ligand. To begin we treated Schrock's complex (Figure 5) with the OCO ligand because of the desired, O-C-O connections. The alkylidene 5 forms after heating the mixture at 85 °C. A manuscript describing this work is in preparation. Based on these preliminary results the PI secured additional funding through the NSF-CAREER program.