

47139-G1
Asymmetric Hydrovinylation and Related Reactions
The goal of this research is to develop new chiral ruthenium catalysts
that will mediate the enantioselective functionalization of prochiral
olefins.
Initial work has been directed toward the development of a catalytic,
enantioselective hydrovinylation reaction. We began this process by
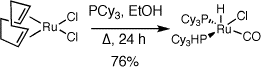
utilizing a ruthenium hydride complex containing two monodentate
phosphine ligands. This complex is easy to synthesize in one step and
can be made readily on multigram scale. Even more importantly, this
complex was able to effectively catalyze hydrovinylation reactions at
low catalyst loadings at room temperature. We can catalyze a number of
efficient reactions, including electron-rich and electron-poor arene
substrates with the same catalyst.
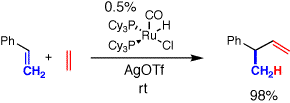
The key to developing the active catalyst was treatment of the
ruthenium chloride precatalyst with silver(I) triflate (AgOTf) to
generate a ruthenium triflate in situ. In some cases the
hexafluoroantimonate (SbF6) anion worked better. This generated an
active catalyst capable of mediating all the desired organic
transformations. Importantly, this catalyst was not overly active, ie
it did not produce by products such as isomers and oligomers that can
be overserved with less selective catalysts. The catalyst operates very
efficiently at room temperature, whereas previous catalysts required
the application of external heat in addition to strong acid to effect
catalyst turnover.
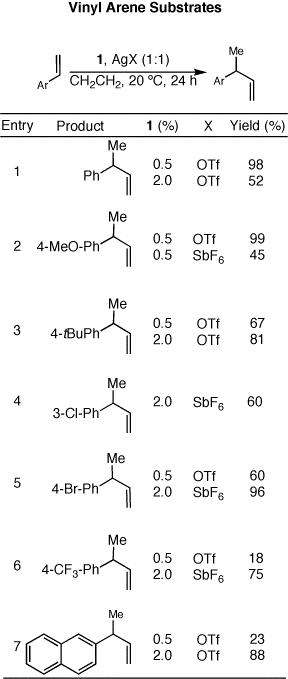
We have noted an interesting catalytic profile for these
ruthenium-based catalysts, notably that they are more effective at low
loadings and low concentrations. This unexpected but beneficial result
has helped us investigate even more efficient catalysts. By taking
advantage of lower catalyst loadings than are normally necessary in
transition metal-catalyzed transformations, we can now very effectively
produce our desired coupled products with only 0.5 mol% of catalyst.
The reasons for this high level of catalytic turnover are currently
under investigation. Preliminary results have indicated that a
bimolecular reaction pathway is responsible for catalyst decomposition
and eventual loss of catalytic activity, so by avoiding bimolecular
reactions, by controlling the concentration of catalyst, we have
effectively increased the turnover number of the catalyst. Notably,
these catalysts are active enough to produce products even at low
concentrations.
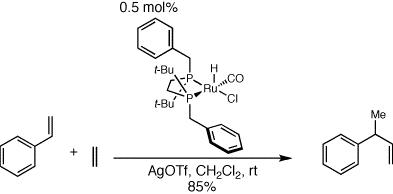
The desireable features described above lead us to continue to develop
this reaction into an enantioselective version. The next step was to
place chelating phosphine ligands on the ruthenium metal center. We
initially opted for aryl phosphine derivatives, but numerous
difficulties led to poor results with these ligands, in both the
complex synthesis and application as a catalyst. We moved our studies
to chelating trialkylphosphines and experienced much better results
with these ligands. In fact, the catalysts derived from these ligands
were almost as active as the monodentate phosphine-derived catalysts
discussed above. The had all the other beneficial features as well,
including ease of synthesis and stability under typical handling and
reaction conditions.
Our current work focuses on introduction of chiral derivatives of these
chelating bidentate phosphine ligands, so that the asymmetric version
of this reaction can finally be realized. This will be the first
asymmetric hydrovinylation reaction catalyzed by a chelating chiral
phosphine ligands. The ligands we are focusing on can be made in 2-3
steps, under typical organic reaction conditions, as we protect the
reactive phosphine with borane. This enables typical workups,
recrystallizations, and flash chromatographic purifications to proceed
without a hitch. The use of catalysts incorporating these chiral
ligands for the hydrovinylation reaction will be the subject of the
next report.