

44206-AC6
The Effect of Conformation and Long-Range Structure on the Femtosecond Dynamics of Electronic Excitation Transfer in Well Defined Polymers: DNA as a Case Study
We are interested in understanding the exciton size and mobility in structured polymer architectures. DNA provides a well-defined structure for exploring the strong electronic interaction between near-degenerate chromophores. We have used native DNA oligomers as model systems where close comparison with theory can be made. This has been supplemented with comparisons to geometrically related aromatic homopolymers. Important factors are whether the exciton hops via random walk before trapping or can find a target trap at greater lengths by coherently sampling large numbers of sites. It is only recently becoming understood that Nature has developed photosynthetic antenna structures that can exploit electronic coherence to overcome inefficient random hopping. Our work has been aimed at determining whether such coherent energy transport pathways are active in double stranded DNA sequences and how the couplings are modulated by conformation of the native double helix. Use of dispersed transient absorption with 30 fs time resolution has allowed us to show the excited state is localized on a dT single strand within our time resolution. This is made possible by our observation of the stimulated emission lineshape at times before the bright character of the excited state has been extinguished. Our results do not support a recent report of a delocalized exciton on (dA)n single strands.
 Our earlier measurements used pump-probe bleach
depolarization to monitor motion of the exciton on double stranded DNA. We
found distinct differences in the depolarization timescale for different
sequences suggesting the exciton (whatever its character) was mobile. It has
been argued by others that in stacked DNA bases, excimer formation (charge
transfer between adjacent bases) takes place very rapidly (< 100 fs)
although the question of mobility of that state has not been established. We
have tackled this key question by several different strategies. On the
theoretical side, we have made calculations of the spectroscopic signature of
excimer states in transient absorption for model stacked aromatic systems and
compared these to CT bands in radical dimer cations. In this way, we can
determine whether the dispersed transient absorption data we (and others) have
can be used to answer the question of whether the mobile state is a weakly
bound excimer or a locally excited state. We have developed an ongoing
collaboration with Eric Bittner (U Houston) to compute the full system
evolution in model DNA sequences allowing for both charge transfer and excited
state migration. On the experimental side, we have successfully compared the
depolarization signatures seen in DNA to those of two model systems that have similar
transition dipole deployments (fanning out in a plane). Two different
macrocycles based on the long-lived fluorene chromophore have allowed us to separate
the effect of p-stacking and large electronic couplings. The first,
poly(9,9-dimethyl-vinyl-fluorene), shows incoherent migration on the 1 ps
timescale and signatures for excimer formation. The second, a conjugated
tetracycle poly(fluorene-3,6-diyl), has no p stacking between
chromophores or charge transfer between units, but conjugation leads to delocalization
over the entire building block in < 500 fs. Laser difficulties during 2007
precluded us finishing an experiment to compare the ultrafast depolarization
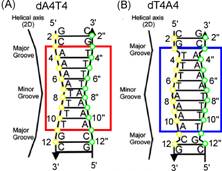
along double stranded A4T4 and T4A4
sequences but we expect this measurement to be complete in the next month. By
comparing oligomers with identical runs of adjacent bases but with opposite
sequence 5'-3' polarity (see figure), we expect to establish the role of conformational
change on electronic couplings and thus exciton mobility.
Our earlier measurements used pump-probe bleach
depolarization to monitor motion of the exciton on double stranded DNA. We
found distinct differences in the depolarization timescale for different
sequences suggesting the exciton (whatever its character) was mobile. It has
been argued by others that in stacked DNA bases, excimer formation (charge
transfer between adjacent bases) takes place very rapidly (< 100 fs)
although the question of mobility of that state has not been established. We
have tackled this key question by several different strategies. On the
theoretical side, we have made calculations of the spectroscopic signature of
excimer states in transient absorption for model stacked aromatic systems and
compared these to CT bands in radical dimer cations. In this way, we can
determine whether the dispersed transient absorption data we (and others) have
can be used to answer the question of whether the mobile state is a weakly
bound excimer or a locally excited state. We have developed an ongoing
collaboration with Eric Bittner (U Houston) to compute the full system
evolution in model DNA sequences allowing for both charge transfer and excited
state migration. On the experimental side, we have successfully compared the
depolarization signatures seen in DNA to those of two model systems that have similar
transition dipole deployments (fanning out in a plane). Two different
macrocycles based on the long-lived fluorene chromophore have allowed us to separate
the effect of p-stacking and large electronic couplings. The first,
poly(9,9-dimethyl-vinyl-fluorene), shows incoherent migration on the 1 ps
timescale and signatures for excimer formation. The second, a conjugated
tetracycle poly(fluorene-3,6-diyl), has no p stacking between
chromophores or charge transfer between units, but conjugation leads to delocalization
over the entire building block in < 500 fs. Laser difficulties during 2007
precluded us finishing an experiment to compare the ultrafast depolarization
along double stranded A4T4 and T4A4
sequences but we expect this measurement to be complete in the next month. By
comparing oligomers with identical runs of adjacent bases but with opposite
sequence 5'-3' polarity (see figure), we expect to establish the role of conformational
change on electronic couplings and thus exciton mobility.
The results of this project have been presented at five seminars and international conferences in Oxford and Paris; the key results are in the process of being published. This project represented a completely new direction in the P.I.'s research. The PRF-funded project has provided important results and insights making possible a five-year proposal currently under consideration with DOE and co-investigator support and collaboration on a successful R01 with NIH. The PRF grant has also had meaningful impact on student training. An undergraduate student from a historically black college was able to participate in summer research. She presented her work at meetings near her home institution (Virginia State) and the 2008 ACS New Orleans meeting. Her advisor was also able to spend six weeks in my laboratory as part of this project. This reconnected him with laser spectroscopy research and allowed him to advise a research student, all of which helped him in his tenure review at VSU. The support of this grant helped one graduate student to completion of his PhD and an opportunity to present his work at the Western Spectroscopy Association meeting. A second student has carried on the project and has also presented at the WSA meeting this year. A small quantum chemistry sub-project by a theory student in my group was also enabled by the PRF grant allowing him to explore excited states of larger aromatic systems.