45992-G3
Tunable Ligands for Rhodium-Catalyzed Hydroformylation
I. Synopsis and Structural Studies
Hydroformylation is elegant in its simplicity – an alkene reacts with CO and H2 to form a synthetically versatile aldehyde with complete atom economy. Even the simplest substrate, a terminal olefin, produces linear (l) and branched (b) aldehydes. The l-aldehyde is typically desired, and we have focused on improving regioselectivity through rational ligand design. The most active mononuclear (pro)catalysts are rhodium complexes of bidentate P-donor ligands. The trigonal bipyramidal complex that functions as the progenitor of the active species can exist as e,e- and e,a-isomers, and the e,e isomer predominantly yields l-aldehydes. We have crystallographically characterized an Ir complex [IrCl(CO)(COD)(L1)], where L1 is the TERPHSPAN diphosphine ligand (Figure 1) and COD is 1,5-cyclooctadiene, in which the TERPHSPAN diphosphine coordinates to Ir in the desired e,e-mode in the pseudo-trigonal bypyramidal complex (a P-Ir-P angle of 105 degrees was observed, Figure 2).
FIGURE 1
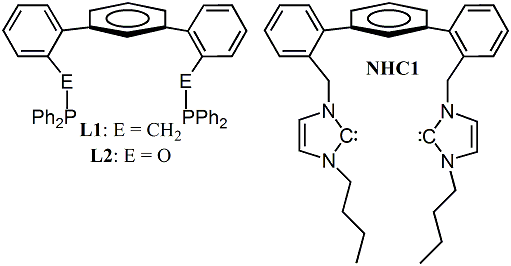
FIGURE 2
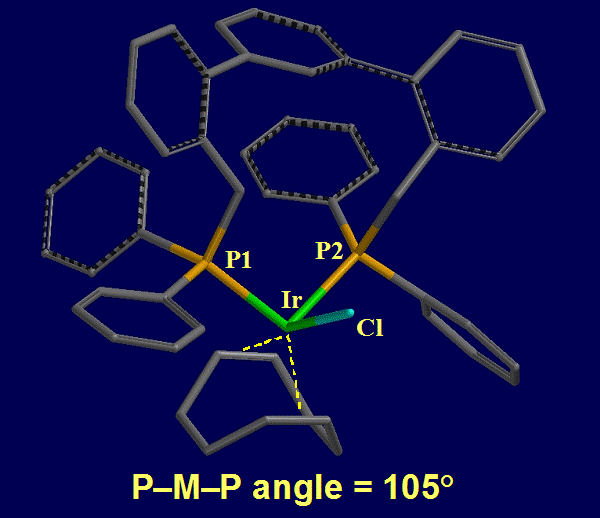
The ability of the ligand to accommodate a trans-spanning mode in square planar complexes (generated from e,e- species during catalysis along the path to the desired regioisomer) is also beneficial. We thus prepared a square planar rhodium complex, [RhCl(CO)(L1)], to confirm this coordination mode. As anticipated, the TERPHSPAN ligand adopts a trans-spanning mode with a P-Rh-P angle of 171 degrees (Figure 3).
FIGURE 3
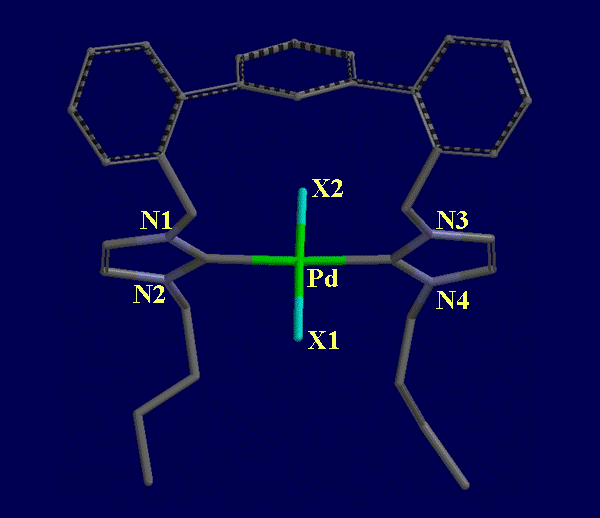
These structural studies were important data points for confirming the results of theoretical calculations we reported on previously. Having demonstrated some favorable geometries for the initial TERPHSPAN ligand (where PR2 = PPh2), we set out to prepare a new set of TERPHSPAN ligands in which phenyl was replaced with ethyl, isopropyl, n-butyl, i-butyl, t-butyl, cyclopentyl, cyclohexyl, o-tolyl, or p-tolyl groups. The aim of preparing this diverse set of diphosphines was to elucidate the electronic and steric effects that such variation would have on the geometry of isolable complexes as well as to probe their effect on catalytic efficiency and regioselectivity. We also replaced the phosphine units of the diphosphine (L1) with phosphinite units (OPPh2, L2) or with N-heterocyclic carbenes (NHC1) so that we could compare the catalytic activity of the three classes of ligands (Ligand structures, Figure 1). Although we have not been able to obtain X-ray structures of complexes with L2, we have obtained a square planar complex, [PdX2(NHC1)] (mixed Cl and Br occupancy at X) featuring the dicarbene ligand in which carbene carbons are observed to coordinate to Pd in a trans-spanning mode similar to that observed in the complex of diphosphine L1, with a C-Pd-C angle of 177 degrees (Figure 4).
FIGURE 4
II. Catalytic C-C Bond-Forming Catalysis utilizing TERPHSPAN ligands
Thus far, we have probed three catalytic reactions using the original TERPHSPAN diphosphines: Rh-catalyzed conjugate addition of arylboronic acids to unsaturated carbonyls; Pd-catalyzed Suzuki-Miyaura coupling of arylboronic acids with aryl halides; and rhodium catalyzed hydroformylation of styrene. Only the hydroformylation results are provided here due to space constraints (Table 1), the other results having been submitted for publication previously. These results are preliminary and are not optimized by any means, but show promise for delivering linear aldehydes even from substrates prone to yielding predominantly branched aldehydes (in this case styrene).
TABLE 1: Preliminary results for hydroformylation of styrene with 1:1 H2/CO gas. S/C is the substrate:catalyst ratio. Ligands are TERPHSPAN diphosphines with R = Ph (1a), o-tolyl (1b), cyclopentyl (1c) and t-butyl (1d).