
Back to Table of Contents
46939-G3
Toward Understanding Biological Nitrogen Fixation: Direct Electrochemical Studies of Nitrogenase
F. Akif Tezcan, University of California, San Diego
After decades of
extensive research, mechanistic details of how nitrogenase accomplishes the
conversion of dinitrogen into ammonia still remain a mystery. Reduction of the
nitrogenase active site metal cluster, FeMoco, is postulated to be required for substrate binding and has only been achieved
under turnover conditions in solution. Consequently, it has been challenging to populate
substrate/intermediate-bound forms of nitrogenase in sufficient quantities for
physical characterization. In order to gain a better understanding of electron
transfer processes within nitrogenase, we had proposed to develop methods for
the direct interrogation of the redox properties of the Fe-S clusters situated
in both nitrogenase components, the Fe-protein (FeP) and the MoFe-protein
(MoFeP). The financial support by the ACS Petroleum Research Fund has
enabled me and my group to pursue this very high risk/payoff line of research
during last year, for which we are extremely grateful. Specifically, these
funds were primarily used for supporting a graduate student, Lauren Roth. It is
due in part to the results she has obtained while being supported by the ACS
PRF G Grant that Lauren was recently awarded an NSF graduate fellowship. Her
efforts during the previous funding period are outlined below.
Despite the
obvious need to establish the redox properties of the nitrogenase Fe-S clusters
and to electronically access them, there are no published reports on their
direct electrochemistry and redox activation, possibly owing to their burial
within the protein medium, particularly in the case of MoFeP. Thus, our efforts
primarily focused on the site-selective labeling of MoFeP with redox- or
photo-active functionalities that would provide conduits for rapid electron
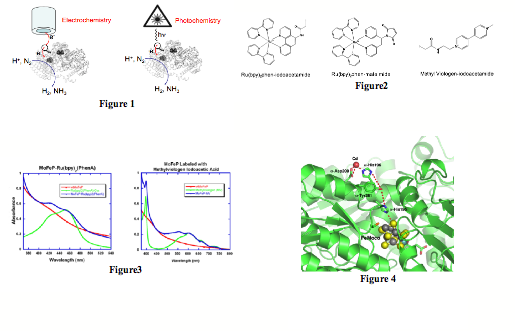
transfer to the buried Fe-S cluster (Fig1). With this goal in mind, we prepared Cys-specific methyl
viologen (MV) and Ru-polypyridine derivatives that could be utilized for
electrochemistry and photochemistry experiments, respectively (Fig2). MoFeP
possesses several Cys residues, but only a few of them (in particular a-Cys45) appear to be sufficiently
solvent-exposed. Fortuitously, Cys45 is close enough to FeMoco (<20Ang) to
allow moderate electronic coupling. A significant fraction of the funding
period was spent on the optimization of the protein labeling conditions and the
determination of the location of functional groups on the protein surface.
These experiments are challenging due to the large size (~250 kDa) of MoFeP, in
addition to the difficulty in chromatographically separating the products from
unlabeled protein. We have succeeded in efficiently labeling MoFeP both with
MV- and Ru-derivatives (Fig3), and have determined that the labels are located
on the a-subunit as intended. The
current efforts focus on the determination of the exact location of chemical
labels on the a-subunit through protein
digestion coupled with mass spectrometry, and X-ray crystallography. We are
also gearing up to determine whether the MV- or Ru-derivatized MoFeP can be
activated for substrate reduction. Our initial experiments will entail the
steady-state photolysis of the Ru-derivatives in the presence of sacrificial
electron donors such as triethanolamine, EDTA or dithionite, followed by GC
detection of H2 evolution. Any detection of H2 over the
background will indicate that FeMoco can be reduced externally under
non-turnover conditions, opening the path to both of the aforementioned goals
of our research program.
In
a simultaneous effort closely coupled to the above experiments, we aim to
delineate proton transfer pathways that lead to FeMoco. Given that each
catalytic cycle in biological nitrogen fixation is coupled to the transfer of 8
protons along with 8 electrons, it is crucial to understand how protons are
delivered to the FeMoco and the substrates. It has been previously shown that
Cd(II) and Zn(II) ions can selectively target residues that form proton entry
points in cytochrome c oxidase and he
bacterial photosynthetic reaction center which also couple proton and electron
transfer reactions. With this in mind, we have determined the structure of
MoFe-protein cocrystallized with Cd(II) (Fig4). The 2.5-ü structure revealed
one Cd(II) ion on the surface each ab-dimer,
coordinated to a-Asp200 and a-His196. These two residues are
directly above FeMoco, and appear to form a proton transfer channel along with
two intervening residues, as postulated earlier by Durrant and others. In order
to probe if this putative proton channel is involved in nitrogenase catalysis,
we performed activity assays in the presence of Cd(II). Cd(II) indeed abolishes
nitrogenase activity, however, we found out that this inactivation takes place
through the destruction of the FeP 4Fe-4S cluster by Cd ions. This finding
further emphasizes the potential importance of electrochemical and
photochemical methods to deliver electrons to FeMoco for substrate activation.
While
the experiments we have carried out in the first funding period have been
slightly different than we have envisioned, they not only form a foundation for
the experiments (direct electrochemistry of nitrogenase) outlined in the
original proposal, but also several other lines of research in nitrogen
fixation, which we are excited to pursue in the upcoming year.
Back to top
