
ACS PRF | ACS
All e-Annual Reports

45062-G6
Quantum Wavepacket Ab Initio Dynamical Studies on Reactivity and Spectroscopy of Hydroxyl and Hydroperoxyl Radicals with Alkenes in Water Clusters
As part of the first year of support from our ACS-PRF-G grant, a total of three papers have been published and a few more are in the process of being completed. Below I briefly highlight our progress from this support.
We started out to study the reaction of hydroxyl radical with isoprene, in a cluster of water molecules, using ab initio molecular dynamics. We soon realized that this problem was compounded by, what we then considered to be, a “rather strange” interaction between the hydroxyl radical and the surrounding water molecules. This development essentially split open this project into two parts and in the paragraphs below I will highlight our progress on each of these multiple fronts.
To ascertain if this rather unexpected dependence of vibrational spectrum on the cluster temperature is unique to hydroxide, we also studied protonated water clusters at a variety of temperatures using ab initio molecular dynamics. We found that the protonated system is less susceptible to such temperature dependent effects in the range of temperatures we probed. We believe the reason for this interesting result is due to the fact that a fluctuating solvation shell (comprising 4-5 water molecules as seen in the figure above) is present for the case of hydroxide ions while the hydronium is prone to display a fixed solvation structure comprising three surrounding water molecules. A brief description of our results has been published in the Journal Chemical Physics. We believe these solvation properties and vibrational properties have a fundamental effect on the reactivity of these ions in the gas-phase due to the abundance of water clusters in the earth's atmosphere. Based on this work, our group has been invited to write a review article on dynamical and temperature dependent effects on vibrational spectroscopy of ionic clusters in the gas-phase. This is the Advances in Quantum Chemistry article listed in the bibliography. With a better understanding of the solvation and cluster temperature dependent vibrational properties of hydroxide in a cluster of water molecules we are currently in the process of probing the flow of energy in small hydroxylated alkenes such as hydroxyl-isoprene and hydroxyl-butadiene, using ab intio molecular dynamics simulations. This flow of energy is expected to have an important bearing on recent experimental results that describe the reaction of hydroxyl radical with alkenes at low temperature conditions. Collaboration with the experimental group of Phil Steven at Indiana University has been initiated and work is ongoing on understanding energy flow. We are currently utilizing instantaneous vibrational density of states, obtained from ab intio dynamics simulations, as a measure of energy flow in such systems. (Interestingly, the importance of vibrational energy flow in these processes was stated by one of our PRF-G proposal reviewers. We are happy to note that we have arrived at a similar conclusion through our independent study.) This should lead to several interesting publications in the very near future. 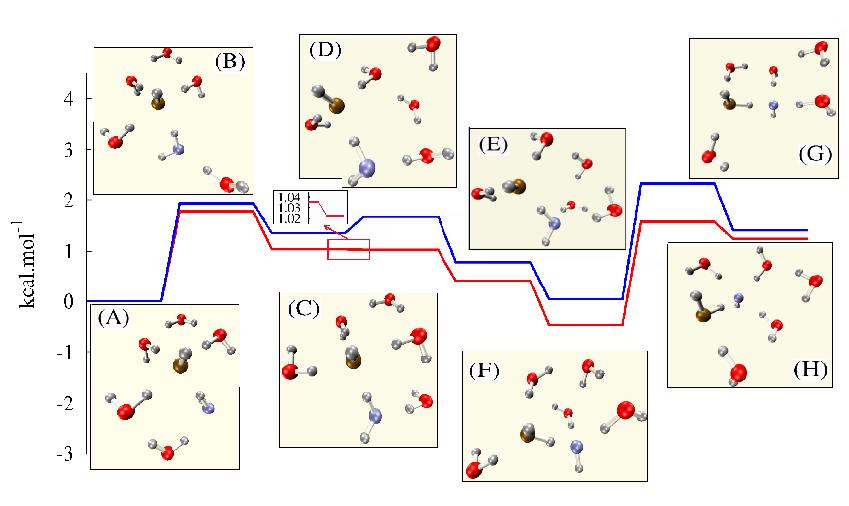 To probe the interaction between the hydroxyl radical and the surrounding water molecules, we were lead by experimental results already available for the hydroxide ion in water. This lead us to a detailed examination of solvation structure, dynamics and vibrational spectrum of a single hydroxide ion surrounded by a few water molecules, under conditions of finite temperature. We, however, found that the hydroxide ion could support a hyper-valent (or five-coordinated) structure in water clusters which could influence its reactivity. Upon probing a cluster of water molecules containing a single hydroxide ion we found the conditions under which a hyper-valent hydroxide ion could be stabilized in a water cluster, which we discussed in a paper now published in the Journal of Physical Chemistry A. The figure beside this paragraph displays a hydroxide hopping mechanism that we have noted in our simulations and shows the propensity of five-coordinated and four-coordinated oxygen atoms in solvated hydroxide systems. In the process of our study we found a critical dependence of the vibrational spectrum on the temperature of the system and we used this idea to predict the conditions under which a penta-valent (or hypervalent) hydroxide ion can be detected using experimental vibrational action spectroscopy.
To probe the interaction between the hydroxyl radical and the surrounding water molecules, we were lead by experimental results already available for the hydroxide ion in water. This lead us to a detailed examination of solvation structure, dynamics and vibrational spectrum of a single hydroxide ion surrounded by a few water molecules, under conditions of finite temperature. We, however, found that the hydroxide ion could support a hyper-valent (or five-coordinated) structure in water clusters which could influence its reactivity. Upon probing a cluster of water molecules containing a single hydroxide ion we found the conditions under which a hyper-valent hydroxide ion could be stabilized in a water cluster, which we discussed in a paper now published in the Journal of Physical Chemistry A. The figure beside this paragraph displays a hydroxide hopping mechanism that we have noted in our simulations and shows the propensity of five-coordinated and four-coordinated oxygen atoms in solvated hydroxide systems. In the process of our study we found a critical dependence of the vibrational spectrum on the temperature of the system and we used this idea to predict the conditions under which a penta-valent (or hypervalent) hydroxide ion can be detected using experimental vibrational action spectroscopy.