
ACS PRF | ACS
All e-Annual Reports

44770-AC3
Heterobimetallic Bismuth-Transition Metal Carboxylates as Catalysts and Catalytic Precursors
The use of tetrabridged dirhodium(II,II) carboxylates, [Rh2(O2CR)4], as catalysts for the reactions of diazo compounds has developed into a powerful tool in synthetic organic chemistry. Dirhodium complexes are known to catalyze a great variety of chemical processes ranging from C-H activation and X-H (X = N, O, Si, S) insertion to cyclopropanation, cyclopropenation, and ylide transformation. The development of dirhodium catalysts has mainly been centered around modification of the bridging carboxylates including their partial or full replacement by other monoanionic ligands.
We have introduced an additional route for modification of the rigid core dirhodium paddlewheel catalysts: formation of a heterobimetallic unit by replacing one of the rhodium atoms with less expensive metal. This idea is based on computational and inhibition kinetic studies of the rhodium-catalyzed carbenoid reactions indicating that the active [Rh2] catalyst uses only one of its two coordination sites at a time for carbene binding. By utilizing the unique ability of bismuth(II) trifluoroacetate (A) to act as a metalloligand toward transition metal fragments, we suggested a preparative solid-state technique that affords heterobimetallic homoleptic carboxylate [BiRh(O2CCF3)4] (B). This remarkable molecule maintains a fully-ordered paddlewheel structure with a single bismuth-rhodium bonding interaction (2.55 Ĺ) as confirmed by theoretical calculations. Further study of the reactivity of bismuth(II) trifluoroacetate has led to the development of an effective one-step synthetic route to heterobimetallic mixed-ligand carboxylates, [cis-BiRh(O2CCF3)2(O2CCMe3)2] (C) and [BiRh(O2CCF3)3(O2CCH3)] (D). These air-stable molecules feature a paddlewheel structure that is retained in solution as well as upon coordination by donor groups. They exhibit an avid one-end Lewis acidity at the rhodium metal site only.
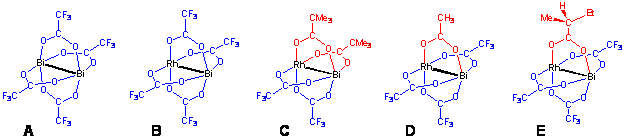
The synthetic method based on solid-state interaction of bismuth(II) trifluoroacetate with unsolvated dirhodium(II) carboxylates allows us to effectively modify the structures of dirhodium catalysts. First, it affords one-end Lewis acids in which the second atom of the heterobimetallic unit does not participate in the coordination of basic substrates but can still act as an electron pool and as an anchor for the bridging ligands. Second, it permits to tune finely the properties of the heterobimetallic molecules, such as thermal stability, moisture and air sensitivity, solubility, and volatility as well as Lewis acidity and steric hindrance about the rhodium axial coordination site. Most importantly, it facilitates the introduction of carboxylate ligands that would bring excellent control of regioselectivity, diastereoselectivity, and enantioselectivity in catalytic reactions. To illustrate the latter, a homochiral heterobimetallic molecule [BiRh(O2CCF3)3(O2CBus)] (E) harvesting one chiral (S)-(+)-2-methylbutyrate ligand has been isolated and characterized. Preliminary tests indicate that heterobimetallic bismuth-rhodium complexes exhibit catalytic activity in the transformations of diazo compounds similar to that of homometallic dirhodium counterparts.