
ACS PRF | ACS
All e-Annual Reports

42180-G1
An Allene-Centered Pericyclic Reaction Sequence for the Synthesis of the Cyathane Diterpenes
The specific research that was the subject of the original proposal is no longer in progress in the group, due to unforeseen difficulties and related work that appeared in the literature. Instead, the PRF funding was used to support similar research in synthetic organic chemistry. We published five papers — two methodology projects and one synthesis — that acknowledge support from the PRF. The two methods address general problems in organic synthetic technology; we intend to draw on both in future work.
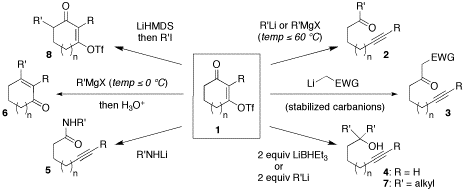
Vinylogous acyl triflates had received little attention beyond the context of transition metal-catalyzed cross-coupling reactions. A thorough analysis of the chemistry of vinylogous acyl triflates provides insight into important chemical processes and opens new directions in synthetic technology. Funding from the PRF arrived in time to support efforts on the full account of our studies.[1] Tandem nucleophilic addition/C–C bond cleaving fragmentation reactions of cyclic vinylogous acyl triflates 1 yield a variety of acyclic acetylenic compounds. These reactions are proposed to proceed through 1,2-addition of the nucleophile to the carbonyl group of triflates 1 to form a tetrahedral alkoxide intermediate, which undergoes a Grob-type C–C bond cleavage to yield acyclic acetylenic compounds 2–5 and 7. The potent nucleofugacity of the triflate moiety is channeled through the sigma-bond framework of 1, providing direct access to the fragmentation pathway without denying other typical reactions of cyclic vinylogous esters. Our study demonstrates the synthetic versatility of vinylogous acyl triflates.

One class of products, alkynyl aldehydes, could not be obtained with reasonable efficiency under our original protocol. Because aldehydes are not stable to the reaction conditions, we then focused on vinylogous triflate hemiacetals, which are isolated in good yield by addition of DIBAL to the carbocyclic vinylogous acyl triflates. Upon reaction with Grignard reagents, these stable tetrahedral intermediates re-enter the fragmentation pathway, generating aldehydes in situ for subsequent addition of the Grignard reagent. Thus, the vinylogous triflate hemiacetals serve as surrogates for labile alkynyl aldehydes.[2] More recently, oxacyclic vinylogous acyl triflates have proven amenable to the same reaction pathway. The important difference is that the resulting products are chiral homopropargylic alcohols. This finding opens up a new avenue of potential applications for our methodology, as we now have an original entry into the synthesis of polyketide building blocks. The second methodology supported by the PRF grant resulted in an atom-economical and efficient olefination strategy for ketones. Ethoxyacetylide addition followed by a gold-catalyzed Meyer–Schuster rearrangement affords a,b-unsaturated esters, generally in excellent overall yield from the starting ketones. The alkynophilicity of Au3+ promotes an interaction with the electron-rich acetylenes that catalyzes the Meyer–Schuster rearrangement selectively over other conceivable pathways.[3]

Compared to many carbanion nucleophiles, acetylides are less basic and less sensitive to steric congestion around the electrophilic carbonyl partner. The idealized sequence of (1) addition of a terminal acetylene and (2) Meyer–Schuster rearrangement results in olefination of the carbonyl with complete atom economy. This two-step strategy is particularly valuable for the Horner–Wadsworth–Emmons (HWE)-type olefination of hindered ketones. It was this potential utility that stimulated our interest in developing a mild and generally efficient protocol for conducting the Meyer–Schuster reaction. Current and future work includes efforts to control the stereoselectivity of the Meyer–Schuster reaction with respect to E- and Z-isomers[4] and to apply this olefination strategy to solve problems in chemical synthesis related to the synthesis of the arteannuins, chemical precursors to the antimalaria drug artemisinin.

We recently completed the synthesis of one important member of the arteannuin family;[5] (+)-dihydro-epi-deoxyarteannuin B is formed alongside artemisinin during the autooxidation of dihydroartemisinic acid. Key steps in our synthesis include the use of zinc enolates and Hoye's relay RCM strategy to overcome steric congestion of the menthone core. This work lays the foundation for our long-term objective of developing efficient synthetic access to endoperoxide antimalarials related to artemisinin.
Our research program is designed to challenge students and further their scientific development as we make tangible strides towards our research goals. Young men and women from diverse backgrounds contribute to these research programs. Our research efforts advance the science of organic chemistry while encouraging and preparing students at all levels to include chemistry research in their future endeavors.