ACS PRF | ACS
All e-Annual Reports

43746-G2
Controls on the Hydrogen Isotopic Composition of Petroleum Hydrocarbons
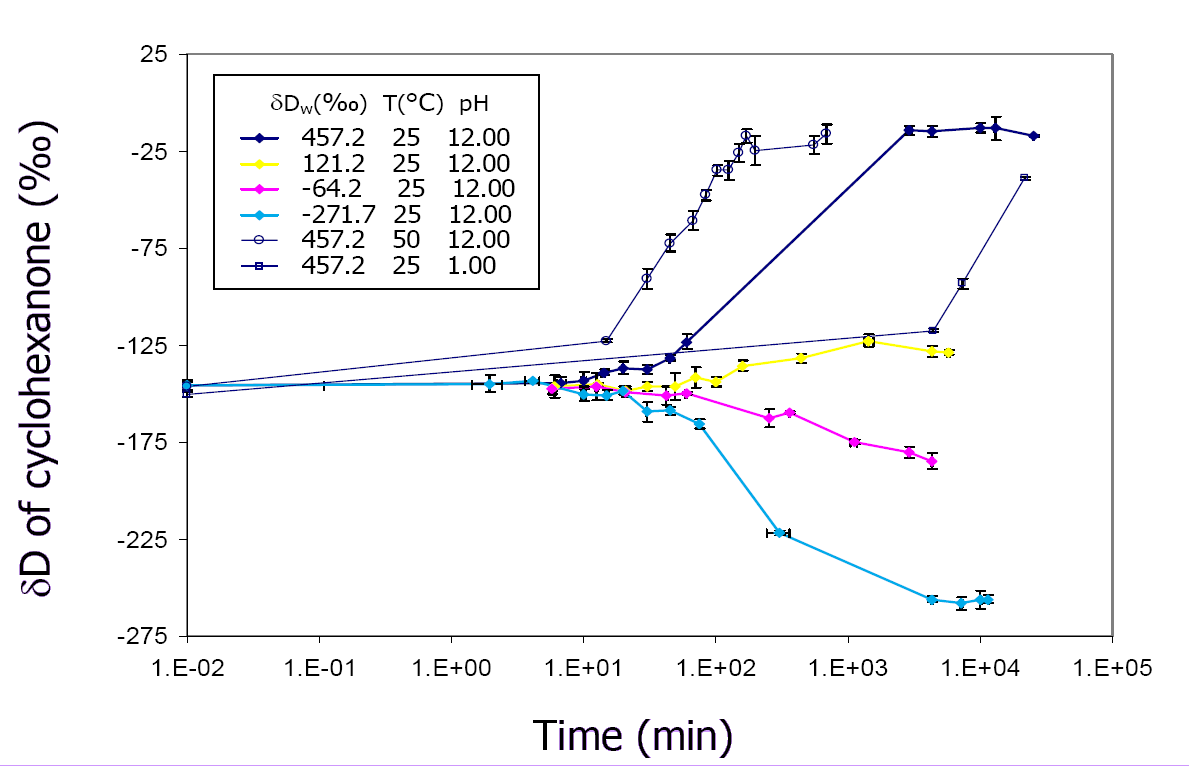
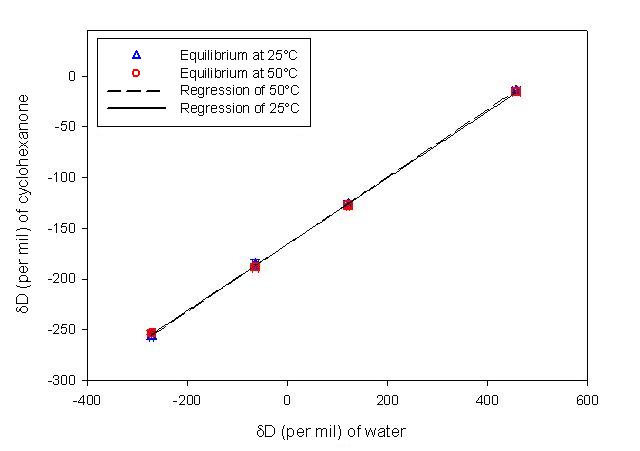
The goal of this project has been to understand the process of hydrogen exchange in organic molecules, and the isotopic (D/H) fractionation accompanying it. Such processes affect even C-bound H over million-year timescales at temperatures above ~80°C, and thus potentially impact the D/H ratios of petroleum hydrocarbons. Because this exchange is so slow, laboratory experiments on simple hydrocarbons are not feasible, even at elevated temperatures. Our approach has been to conduct exchange experiments on simple ketones, taking advantage of rapid keto-enol tautomerism under base catalysis to achieve equilibration of the adjacent (a) hydrogens over several days. Several parallel experiments use water with a systematically varying D/H ratio, in order to correct for non-exchanging H. At selected intervals a sealed ampule is opened and the D/H ratios of both organic analyte and water are measured. The increase in D content of the organic molecule over time provides the kinetic rate constant for the exchange (Fig 1), while comparison of the final D concentrations in analyte and water provides the equilibrium fractionation factor (Fig 2). Our preliminary results for cyclohexanone indicate that this fractionation is -147±9‰ and -150±7‰ at 50° and 70°C, respectively.
The experimental approach can thus far only be used to directly measure H positions adjacent to carbonyl functions. To extend our results to other molecular positions, and to other hydrocarbon molecules, we used ab initio molecular modeling. Calculation of vibrational frequencies was performed with the B3LYP/6-311g** basis set using Jaguar 6.5. The solvent effect was introduced as the electrostatic influence of a continuous dielectric medium. Reduced partition functions of organic molecules are calculated directly, while those of liquid water are obtained by combining gas phase calculations with experimental liquid-vapor fractionation factors. Using our experimental data for cyclohexanone as calibration, we estimate fractionation factors for n-alkyl H in the range of -108‰ at 25°C to -129‰ at 90°C. These predicted hydrocarbon-water fractionations are very similar to those observed in mature petroleum systems.
One unexpected outcome of this project has been a more complete understanding of processes affecting accuracy and precision in GC-pyr-IRMS. Because our experiments require very accurate measurement of organic D/H over a large range, we synthesized multiple organic standards having dD values ranging from -200 to +800‰. Careful manipulation of the order and D/H ratio of these standards allows us to show that there is a small, systematic memory effect in GC-pyr-IRMS systems, such that 2-4% of the hydrogen in each chromatographic peak is derived from that in the preceding peak. In experiments such as ours, where the desired results are obtained by varying D/H ratios over hundreds of permil, this memory can introduce a systematic bias of 20-40‰ in fractionation factors unless properly accounted for.
Figure 1 (left). Kinetic profile of D incorporation into cyclohexanone over time. Different curves represent experiments using water with differing D/H ratios. Figure 2 (right). Plot of the equilibrium D/H ratios of organic analyte and water. The fractionation factor is derived from the slope of the regression