ACS PRF | ACS
All e-Annual Reports

45648-AC3
Lewis Acid Chemistry at the Edge of Ferrocene
Bidentate Lewis acids have attracted much recent interest due to their superior performance in diverse areas ranging from Lewis acid catalysis of organic and organometallic reactions, activation of catalysts in olefin polymerization, selective and efficient molecular recognition of anions and neutral nucleophiles, to new electronically interesting materials. The purpose of the research under this grant was to explore the chemistry of ferrocenes with two adjacent Lewis acid centers attached to one of the cyclopentadienyl rings. While the more easily accessible 1,1'-dimetalated ferrocenes have been widely studied in the past, the chemistry of the related 1,2-difunctionalized ferrocenes is virtually unexplored. The latter are of particular interest since they represent three-dimensional, redox-active analogs of the well-established and highly useful bidentate Lewis acids with phenylene and naphthalene backbones.
During the current grant period we have
(A) studied the binding of fluoride anions to heteronuclear bidentate Lewis acids that feature Lewis acidic organotin and organoboron moieties adjacent to one another at the edge of ferrocene, and
(B) investigated the electronic structure of diboradiferrocenes, in which two adjacent Lewis acidic organoboron moieties lead to a doubly-bridged diferrocene system. Particular emphasis was placed on the investigation of electronic interactions in the mixed-valent monocation, and the effect of ferrocene oxidation on the Lewis acidity of the boron centers.
(A) Fluoride Binding to Heteronuclear Bidentate Lewis Acids.
A series of m2-fluoro bridged heteronuclear bidentate Lewis acid complexes was prepared. The coordination of the anion affects both, the boron and tin centers, pyramidalizing the former while imposing pseudo-trigonal bipyramidal geometry on the latter. Solid state and solution data indicate stronger binding of the fluoride anion to the boron center and only weak to moderate interactions with the tin center. A careful comparison of the individual compounds with respect to their structural parameters from X-ray analysis and their NMR spectroscopic data, in particular an evaluation of the 119Sn-19F coupling constants, indicates relatively stronger SnÉF interactions for complexes with R=Ph in comparison to R=F. This suggests that a truly bridging situation of fluoride is favored when the Lewis acidity of boron is diminished and hence more comparable to the strength of the organotin moiety.
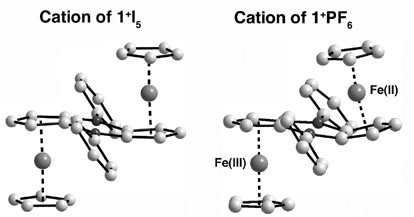
(B) Investigation of the Mixed Valence State of Diboradiferrocenes. The mixed-valent complexes [Cp2Fe2(m-C10H6(BPh)2)]+X ([1+]X; X = I5, PF6, SbF6, B(C6F5)4) were prepared by oxidation of the diboradiferrocene [Cp2Fe2(m-C10H6(BPh)2)] (1) with I2, AgPF6, and AgSbF6, respectively, and in the case of X = B(C6F5)4 through anion exchange of the I5- salt with [Li(Et2O)x][B(C6F5)4]. Investigation of the mixed-valence state of 1 in solution by near IR spectroscopy provided evidence of broad NIR absorptions that are consistent with a Class II mixed-valent cation, where fast electron hopping occurs between two essentially localized states. A comparison to other systems with saturated dimethylsilicon and unsaturated vinylene and acetylene bridges suggests that the rigid double-bridge featuring two tricoordinate boron centers more effectively promotes electronic interactions. The exceptionally large redox splitting of DE = 510 mV in the cyclic voltammogram further supports this view. The question of electronic delocalization in the solid state has been addressed by single crystal X-ray diffraction and Mšssbauer studies. Inversion symmetry has previously been demonstrated to be critical for fast electron transfer to occur in mixed-valent diferrocene cations. In our case, inversion symmetry was found with X = I5 and B(C6F5)4 as the counteranion, which should favor electron transfer. However, for [1+]X (X= PF6, SbF6) the bridging boryl groups are strongly bent towards the iron center of the neutral ferrocene moiety, but much less so to the oxidized ferrocenium moiety. Indeed, Mšssbauer data that were obtained in collaboration with the Herber group show fast electron transfer only with X = I5 and B(C6F5)4 in the temperature range studied. Fig. 1 Comparison of the structures of [1+]I5 (centrosymmetric) and [1+]PF6 (non-centrosymmetric). Finally, we found that the Lewis acidity of boron in diboradiferrocene 1 is strongly enhanced through oxidation of the iron atoms as evident from examination of X-ray structural parameters of the mixed-valent cation [1+]PF6 and further confirmed by the strong donor complexation to the dication obtained upon oxidation in the presence of a donor solvent (acetonitrile, pyridine). These observations suggest the utility of 1 as a powerful Lewis acid in applications ranging from anion recognition to Lewis acid catalysis. These studies are currently under way.