www.acsprf.org
Reports: ND549221-ND5: PGM-Substituted Complex Oxide Catalysts for Selective Hydrocarbon Transformations
Metal oxides, dispersed metal cations associated with the support, small metal particles, and isolated zero-valent metal atoms, have all be proposed as responsible for catalytic activity in simple reaction systems, such as the oxidation of CO and CH4. Finding supports that stabilize the appropriate metal sites (e.g., valence, nuclearity) is critical to optimizing and preserving their activity, and consequently to minimizing the use of precious metals in catalytic hydrocarbon transformations.
Crystalline complex oxides containing precious metal cations at well-defined positions may be useful as mechanistic probes for the roles of such cations in catalysis, if they are very robust under the entire range and duration of reaction conditions (i.e., if the catalytically active metal sites do not change form). Initially, we studied Pd-doped oxides in the family Y(Fe,Mn)O3, containing up to 10 at% Pd on the transition metal site. While there was concrete evidence of reversible Pd egress from the host oxide under reducing conditions (5 %H2, ³ 200 °C) and ingress under oxidizing conditions (O2, 800 °C), the Pd K-edge EXAFS was unable to distinguish the coordination environment of Pd in the complex oxide host from PdO, and the CO oxidation activity was not reproducible. These observations led us to conclude that this complex oxide host is incapable of sufficiently stabilizing cationic Pd. Therefore we sought a more robust family of materials in which Pd was not a dopant, but a stoichiometric constituent.
We prepared a series of oxides Ln2BaPdO5 (Ln = La, Nd, Gd, Dy), by a high temperature solid-state synthesis method, giving materials with low BET surface areas (ca. 1 m2/g). Their structures were investigated by powder X-ray diffraction (XRD) and X-ray absorption spectroscopy (XAS). The XAS results are shown for Ln = Dy in Figure 1. Each oxides contains Pd as square-planar [PdIIO4], incorporated into the complex oxide structure, Figure 2.

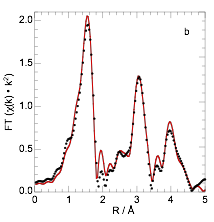
Figure 1. Pd K-edge EXAFS for Dy2BaPdO5, in (a) R-space; and (b) k2-weighted k-space. (Black circles are data, red lines are curvefits to the structure shown in Figure 2).
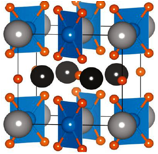
Figure 2. General structure of Ln2BaPdO5. Key: Pd (blue), O (orange), Ln (grey), Ba (black).
To demonstrate their robustness, we recorded lightoff profiles for CO oxidation (1000 ppm CO, 10 vol% O2, balance Ar, 50 mL/min) over each oxide in a packed bed reactor. Representative results are shown in Figure 3a for 15 mg Nd2BaPdO5, diluted with an equal mass of cordierite to avoid hot spots. Provided the catalyst is preconditioned in the reactive atmospheres, consecutive profiles are perfectly superimposable, showing that there is no change in structure during the reaction. Consequently, the lightoff profiles can be analyzed by curvefitting, using the integrated form of the packed bed reactor design equation and the appropriate rate law. Mass transfer limitations are unimportant, since the catalyst is non-porous. In the case of CO oxidation under CO-lean conditions, the reaction is inverse-first-order in P(CO). The resulting activation energy, (94.9 ± 0.1) kcal/mol, agrees with the value determined by the conventional Arrhenius method (Figure 3b), although it can be determined much more precisely from the non-isothermal data.


Figure 3. (a) Consecutive lightoff profiles for CO oxidation catalyzed by Nd2BaPdO5, under CO-lean conditions; and (b) Arrhenius treatment of isothermal data obtained using the same catalyst.
XPS analysis showed no change in the region of the Pd 3d spin-orbit doublet before and after reaction, confirming the absence of metallic Pd under CO-lean reaction conditions, and thereby identifying the active sites as Pd(II). However, under CO-rich conditions, the rate law changes to become zero-order in CO and the XPS shows evidence for near-surface Pd(0). These results suggest that, despite the considerable stability of the complex oxide, the removal of lattice oxygen can occur under reaction conditions, causing reduction of Pd(II).