www.acsprf.org
Reports: ND449785-ND4: Topologically Diverse New Polycyclic Scaffolds via Key Photochemical Steps
The proposed research is to develop a series of efficient synthetic pathways to topologically diverse polycyclic and polyheterocyclic scaffolds by combining rationally selected thermal and photoinduced cycloaddition reactions. In the first year we primarily focused our research on the first direction (same numbering of the aims as in the original proposal): Ci-symmetric and mismatched polycyclic scaffolds derived from tandem double Diels-Alderdouble Paternò-Büchi (P.-B.) cycloadditions.
We have investigated the scope and synthetic potential of this modular synthetic approach, which is based on harvesting the strain installed in a key photochemical step via carefully selected ground state reactions offering access to highly unusual polycyclic scaffolds.
We first studied the D.-A.
adducts of 2-cyclohexenone and 2-cycloheptenone, and found that they are indeed
capable of forming strained polycyclic oxetanes 5-8 upon irradiation (Scheme
1). Strikingly, even the bis-adduct of
cyclohexadiene and benzoquinone 9
was found to be photoactive, producing C2-symmetric dioxetane 11 via the monooxetane 10 when irradiated (the structures of
many strained polycycles described in this report, including the bis-oxetane 11, were unambiguously determined by
XRay crystallography).Scheme 1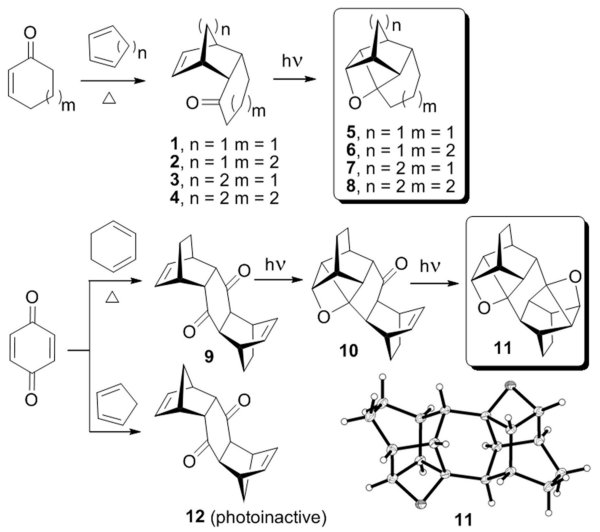
In the bis-series, the photoreactivity limit is reached with the cyclopentadiene adduct 12, which does not form even a monooxetane, unlike the similarly sized cyclohexano-norbornene 1, which is photoactive. This further confirms that flexibility of the ketone-containing ring is critical.
These polycyclic oxetanes are significantly strained and readily undergo acid-catalyzed ring opening and subsequent cationic rearrangements, conforming to certain computationally predictable topologies consistent with the structure of the initial polycyclic enone (Scheme 2).
Scheme 2.
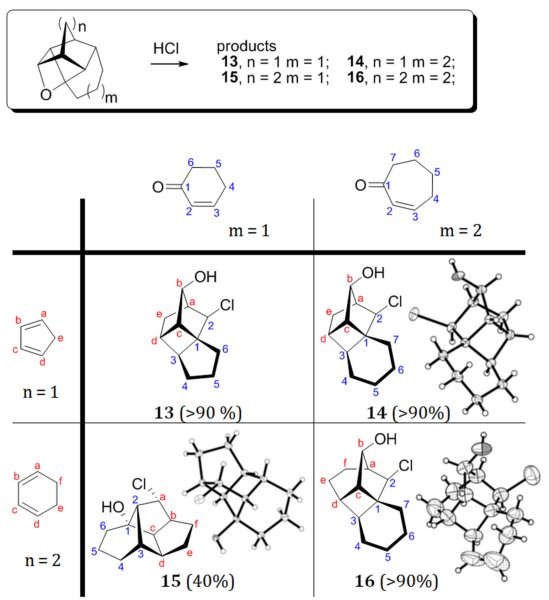
When treated with HCl, oxetanes usually produce 1,3-chlorohydrins. However, in the case of strained oxetanes 5-8,11, which can potentially open to form both tertiary or secondary carbocations, the produced chlorohydrins resulted from more elaborate cationic rearrangements.
Relative stability of the tertiary cation depends on the degree of its pyramidalization, which in these systems is defined primarily by the size of the enone ring. Our rationale is that for larger rings (m>1) the acid-catalyzed oxetane ring-opening preferentially produces the expected tertiary cation of type A1, Scheme 3A. While relatively more stable than the alternative secondary cation, A1 is still destabilized by forced pyramidalization. It relaxes by (remarkably) rearranging into A2 possessing a cyclobutyl moiety. A2 is then trapped by the chloride anion to yield the observed products 13-14, 16.
Scheme 3.
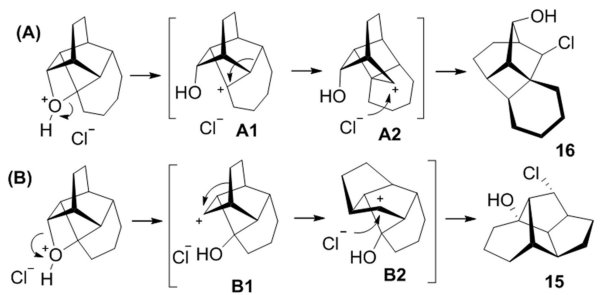
We note that the stereochemical outcome of this two step rearrangement is inversion at each step, which implies that the whole sequence might be a concerted reaction or nearly so.
For m = 1, i.e. the smallest ring size at which Paternò-Büchi cyclization is possible in this series, the tertiary cation is even more destabilized. It is plausible that the two cations are formed reversibly and competitively. The outcome in this case depends on n, i.e. the size of the top bridge. With n = 2, Scheme 3B, upon formation of a secondary cation B1, the two carbon bridge undergoes 1,2-shift to give B2, which is trapped by the chloride anion. Again, all steps in the B-sequence occur with inversion of configuration, which hints at a semi-concerted mechanism.
In the case when both m = 1 and n = 1 as in 5, the single methylene bridge fails to migrate and pathway A prevails, yielding cyclobutane 13.
As it is clearly demonstrated, protolytic heterolysis of a C-O bond in oxetanes 5-8 provides insufficient relief of strain installed at the photochemical step, and therefore subsequent carbocationic rearrangements accompany the oxetane ring opening.
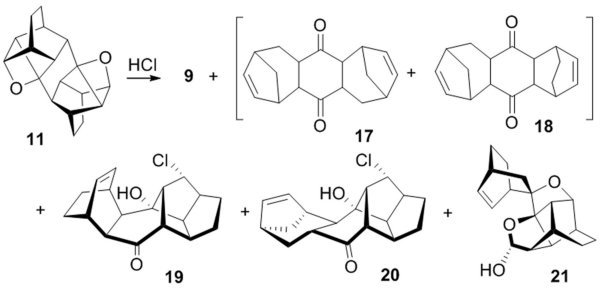
The polycyclic oxetanes are very reactive; they do not generally survive column chromatography. This is especially true for bis-oxetane 11. We found that unlike oxetanes 5-8, bis-oxetane 11 undergoes a number of competing ring-openings and rearrangements under acid-catalyzed conditions, Scheme 4. The major product is an isomer of the starting diene 9 for which we tentatively assign structure 17. The rearranged bicyclo[3.2.1] moiety of 17 (and of its minor isomer 18) is inferred from the NMR data and by analogy with the XRay structure of chlorohydrin 20 which has this moiety. Structures of chlorohydrin 19 and hemiacetal 21 were also elucidated by X-ray crystallography.
It appears that bis-oxetane 11, as most extremely strained, offers a variety of deep skeletal rearrangments under protolytic ring opening conditions. We note, however, that perhaps for this very reason the selectivity and reproducibility of the reaction shown in Scheme 4 was poor. In several runs alkene 17 was forming at 40-50% yield, whereas the yields of products 18-21 never exceeded 10%.
Chlorohydrins 19 and 20 are expectedly related to product 15, which further confirms the generality of the rearrangement shown in Scheme 3B for the oxetanes derived from six membered dienes and dienophiles.
Scheme 4.
In summary, strained polycyclic oxetanes can be synthesized photochemically from the D.-A. adducts of cyclic dienes and enones generally as long as the dienophile has a ring size greater than five. Such oxetanes harvest and store excited state energy, which can be utilized to access unique functionalized polycyclic scaffolds via acid-catalyzed oxetane ring opening and subsequent deep cationic rearrangements.
We are now expanding this methodology onto aliphatic and aromatic ketones. Also, in the second year of studies we are focusing on the photochemistry of the starting materials obtained from hetero-Diels-Alder cycloadditions, primarily the thiocarbonyl compounds. The initial results are encouraging, as we get expeditious access to bicyclo[3.3.0] cores (cyclopenteno-dihydrofurans).