www.acsprf.org
Reports: DNI149072-DNI1: Ligand-Linked Catalysis: Metal-Catalyzed Functionalization via Transient Directing Group Installation
Introduction. Through the invention of synthetic reactions differential from currently existing technologies, we may further enable novel, highly efficient approaches to desired molecular structures. Our funded proposal outlined a general approach to the site-selective functionalization of relatively unreactive bonds by transition metal catalysts. This proposal represented a unification of two distinct concepts: the metal-catalyzed functionalization of unreactive bonds, and the reversible installation of a ligating species that can render a process intramolecular. In this approach, the ligand will act as the link between substrate and catalyst that allows and directs the desired reaction to occur (Figure 1). It is envisioned that an equilibrating transacetalization process between the substrate and the organic molecule catalyst will form an intermediate in situ. This covalently attached substrate-ligand adduct will then direct (via a ligating group Lig) the transition metal to functionalize the substrate in a specific fashion, which upon hydrolytic release of the organic catalyst will afford the product. It is anticipated that this versatile strategy to functionalization will lead to a variety of transformations currently unrealized by either transition metal catalysis or organocatalysis approaches. Figure 1. Metal-Catalyzed Functionalization of Aldehyde-Based Substrates via Transient Covalent Attachment.

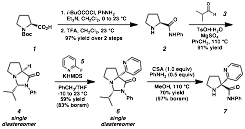
Progress. Our investigations began targeting the design of a specific molecular framework that could enable functionalization in the desired fashion. Two design elements were critical in the formulation of the molecular scaffold: 1) an appropriately positioned directing group capable of ligating metals, and 2) a functional group capable of ultimately transient covalent attachment. The synthesis of our amino amide scaffold, derived from proline, is illustrated in Scheme 1. As can be seen, the installation of a pyridyl ring via nucleophilic aromatic substitution, provides a suitable ligating group for the desired metal-catalyzed functionalizations.
Scheme 1
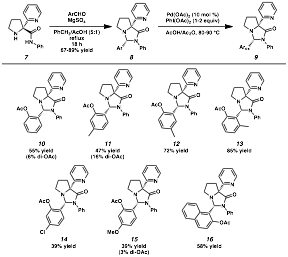
The molecular scaffold can be readily attached to aldehydes to enable directed palladium-catalyzed acetoxylations (Scheme 2). Treatment of the amino amide (7) and an aromatic aldehyde under acetalization conditions affords the corresponding aminals. When each aminal was subjected to oxidation, functionalization at the ortho position of the arene was observed. The compounds were obtained in moderate to good yields overall for a variety of aromatic compounds, with small amounts of diacetoxylation observed in a few cases.
Scheme 2
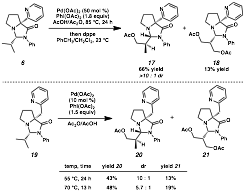
In addition to the functionalization of C(sp2)–H bonds, we investigated the analogous reactions of C(sp3)–H bonds. The same scaffold, as well as a one carbon homolog synthesized in the same fashion, can be bound to isobutyraldehyde in aminals 6 and 19, respectively (Scheme 3). Subjecting aminal 6 to oxidative palladium catalysis produced acetate 17 predominantly. The oxidation occurred with nearly singular site-selectivity, where the molecular framework directs the functionalization to primarily one carbon. The catalyst loadings were quite high; we presently attribute this requirement to the low dissociation capability of the ligand-metal complex. Aminal 19, with the directing group possessing additional conformational flexibility, was treated with similar oxidative conditions. In this system, the relative reactivity was much greater, with oxidation occurring at lower catalyst loadings and decreased reaction temperatures. Monoacetate 20 was predominantly produced, although the minor diastereomer was observed, consistent with the increased flexibility of the scaffold. Selectivities were still quite high, however, reaching 10:1 dr at 55 °C. Importantly, these outcomes establish the capability of this scaffold for site-specific oxidation, and these unique modes of selectivity hold promise for future novel transformations.
Scheme 3
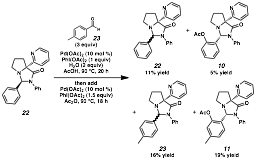
Referring back to Figure 1, we are envisioning systems that have the potential to be catalytic in both metal and organic framework. In order for this goal to be reached, we must achieve compatibility between functionalization and transacetalization. Specifically in these systems, the following must occur starting with an aminal based on our scaffold: 1) hydrolysis to release the amino amide, 2) acetalization with an aldehydic compound, and 3) oxidative functionalization. In the event, we have treated aminal 22 and p-tolualdehyde under the oxidative conditions with added water to assist in exchange (Scheme 4). Under these conditions, we have observed aminals derived from both benzaldehyde and tolualdehyde, as well as their corresponding oxidation products 10 and 11. Albeit only an initial lead, this outcome establishes the feasibility of this process for catalytic remote functionalization, which we anticipate with further studies will provide entries into new reaction manifolds.
Scheme 4
Summary and Future Outlook. We expect that this type of transformation, once fully developed, could be widely applicable in oxidative functionalization reactions. We plan to investigate the potential of this reactivity in a number of different oxidative manifolds. We also anticipate this concept to be applicable in hydroarylation and hydrovinylation reactions, as well as in C–C bond insertion processes. Ultimately, we have initiated this project with the overarching goal of developing synthetic transformations that possess orthogonal characteristics to those currently in our synthetic toolbox. We believe that our efforts described herein have established a foundation toward that goal.
Our present work in this area has undoubtedly caught the attention of the organic and organometallic chemistry communities. I have presented some of this work at universities and at two oral presentations at summer Gordon Conferences. We have submitted a manuscript detailing these efforts that is currently under revision for publication in Chemical Science. Erin Stache, the graduate student primarily contributing to this project, has recently presented a poster at the National ACS meeting in Denver last month. I believe that the outcomes of our efforts, directly from the support of the ACS grant, has had a measurable impact on our career advancement.