www.acsprf.org
Reports: DNI150491-DNI1: [3,3]-Rearrangements of O-Vinyl Oximes: Stereoselective Synthesis of 1,4-Dicarbonyl Compounds
The specific aim of the proposed project was to develop the [3,3] rearrangement reactivity of O-vinyl oximes as a route to stereochemically defined 1,4-dicarbonyl compounds. The overall goal of the program is to explore and develop the chemistry of O-vinyl oximes as valuable precursors to privileged synthetic intermediates to streamline syntheses of pharmaceutically active molecules and desirable new materials. The specific aim of the project built on our preliminary studies that showed that a variety of O-propenyl oximes are easily accessible from O-allyl oximes via an iridium-catalyzed isomerization reaction and that O-propenyl oximes can be subsequently thermally converted to pyrroles. During the first year of the funding period, we have: 1) discovered that O-vinyl oximes undergo both [1,3] and [3,3] rearrangements and that this reactivity can be controlled by substituent effects and reaction conditions to provide regioselective access to either 2,3,4- or 2,3,5-trisubstituted pyrroles; 2) exploited our discovery of the [1,3] rearrangement of O-vinyl oximes to stereoselectively prepare a-imino ketones and aldehydes; 3) studied the mechanism of the [1,3] rearrangement of O-vinyl oximes; and 4) developed a new method for the preparation of O-vinyl oximes which has provided a facile, general, modular, and stereocontrolled route to our required starting materials.
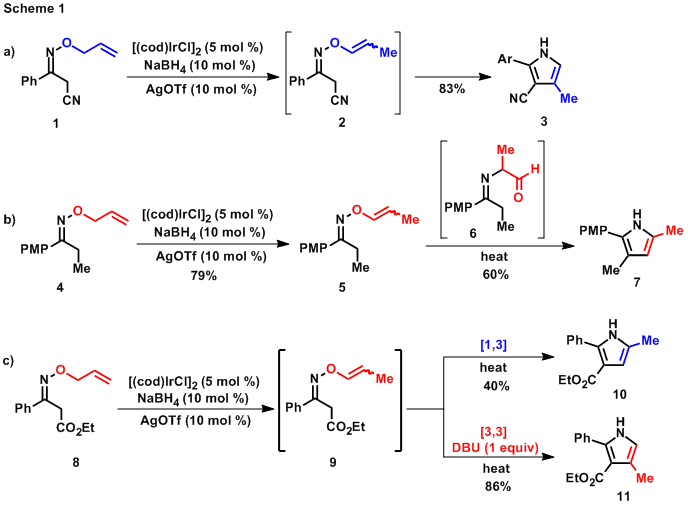
While studying the isomerization of O-allyl oximes and the subsequent rearrangement reactivity of the O-propenyl oxime products, we observed that both the rate of the rearrangement and the regioselectivity of pyrrole formation were dependent on the substitution pattern of the oxime. When a-cyano O-allyl oxime 1 was subjected to isomerization conditions, the corresponding 4-methyl pyrrole 3 was isolated instead of the expected O-vinyl oxime 2 (Scheme 1a). This suggested that the tautomerization, [3,3] rearrangement, and Paal-Knorr cyclization sequence necessary to form the pyrrole was particularly facile for cyano-substituted oximes under the isomerization reaction conditions. In contrast, when isolated alkyl- or aryl-substituted O-vinyl oximes, such as 5, were heated in dioxane, a thermal rearrangement and cyclization was observed to provide 5-methyl pyrrole 7. This change in regioselectivity was not affected by the presence of iridium and was determined by observation of a-imino aldehyde intermediate 6 to be caused by a change in mechanism to a [1,3] rearrangement followed by enamine addition and condensation. The difference between the two mechanistic pathways is most likely caused by the thermodynamic preference of the substrate for tautomerization. In an effort to exploit these observations, we also determined that the addition of DBU to an O-vinyl oxime rearrangement reaction mixture will reverse the regioselectivity preference of pyrrole formation for both b-ester oximes and b-aryl oximes (Scheme 1c). We reported 20 examples for which either 2,3,4- or 2,3,5-trisubstituted pyrroles can be accessed in two steps from the same commercially available starting materials using simply the presence or the absence of DBU in the rearrangement reaction conditions.
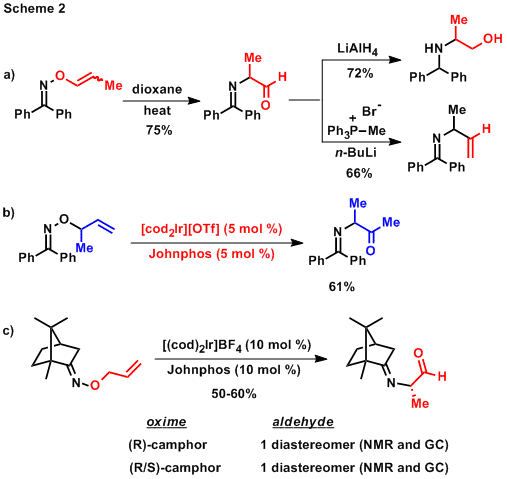
Observation of the [1,3] rearrangement of O-vinyl oximes prompted us to consider O-vinyl oximes as precursors to a-imino carbonyl compounds, which can potentially be converted to a-amino aldehydes or ketones by simple hydrolysis. Non-enolizable O-vinyl oximes were tested to prevent pyrrole formation and the a-imino aldehyde products were derivitized and isolated as amino alcohols and N-allylic imines (Scheme 2a). To directly isolate [1,3] rearrangement products, a-imino ketones were prepared using newly optimized conditions for the isomerization and rearrangement of a-methyl O-allyl oximes (Scheme 2b). The same conditions were tested with enolizable O-allyl oximes and provided access to tetrasubstituted pyrroles as well as the isomerization and rearrangement products of camphor-derived O-allyl oximes. We were particularly excited about the later result and the potential of using camphor as a chiral auxiliary for setting a-amino carbonyl stereocenters. As shown in Scheme 2c, these transformations appear to be stereospecific and will be a major focus of our program during the second year of funding. The stereospecificity of the transformation suggests that the [1,3] rearrangement does not occur via dissociated intermediates. This inference is further supported by Eyring data that we have collected for the [1,3] rearrangement of O-propenyl benzophenone oximes which give a DS value of 3.1 eu. These intitial studies on the [1,3] rearrangement of O-vinyl oximes suggest that this transformation is valuable not only for the regioselective formation of pyrroles but also for the preparation of new carbamine stereocenters.
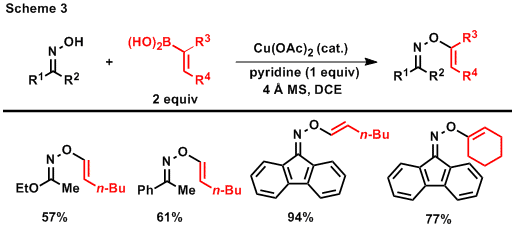
As we began to investigate a wider range of substrates to explore the applications of the [1,3] and [3,3] rearrangements of O-vinyl oximes, our expanding program required a more general method for the preparation of these substrates with a variety of vinyl substitution patterns and specific alkene stereochemistry. To achieve this goal, we decided to pursue copper-catalyzed vinyl etherification in analogy to a report from the Sharpless group which showed that aryl etherification of phthalimide can be achieved with aryl boronic acids and a copper-catalyst.1 Gratifyingly, we have now shown that this transformation is general for the etherification of phthalimide, oximes, and hydroximates, with vinyl boronic acids (Scheme 3). Access to a variety of vinyl substitution patterns is currently allowing us to systematically observe trends in the thermal rearrangement reactions of O-vinyl oximes and evaluate potential catalytic systems. In addition, these copper-catalyzed etherification reactions have brought us closer to our intial goals outlined in the original proposal because the vinyl etherification reaction also tolerates the divinylation of t-butyl hydroxycarbamate. This procedure will now allow us to access divinyl hydroxcarbamates that should undergo facile [3,3] rearrangements and disfavor a Paal-Knorr cyclization to allow for the isolation of 1,4-imino ketones.
In the second year of funding, we plan to use our new method for the preparation of O-vinyl oximes to expand our investigation of using the [1,3] and [3,3] rearrangements of O-vinyl oximes to form tetrasubstituted pyrroles, stereodefined a-imino and a-amino carbonyl compounds, and 1,4-imino ketones.
1. Petrassi, H. M.; Sharpless, K. B.; Kelly, J. W. Org. Lett. 2001, 3, 139.