www.acsprf.org
Reports: DNI350460-DNI3: Low-Valent Uranium Complexes with Redox-Active Ligands for C-O Bond Activation
Biomass is a useful renewable fuel alternative, however, the high oxygen content can cause thermal instability and increase corrosive properties of the fuel. To make biomass a superior fuel alternative, the strong molecular framework must be broken down by activation of the C-O bonds which holds these molecules together. Although transition metals have been widely studied for C-O bond activation processes, the large atomic radius, highly reducing nature, abundance, and low cost of uranium warrant its study for C-O activation and deoxygenation of substrates, including small molecule biomass mimics. Herein, we report our progress towards the synthesis and use of low-valent uranium complexes for activation of small molecule biomass mimics. Our research makes use of redox-active ligands to store reducing equivalents which can be used to break C-O bonds.
Our previous studies of hydrotris(pyrazolyl)borate frameworks have demonstrated that these ligands can be labile, thus the reactivity of similar more robust cyclopentadienyl systems was explored. Reduction of Cp*2UI(THF) (1) in the presence of 2,2-bipyridine using an equivalent of KC8 affords Cp*2U(2,2'-bpy) (2) (Cp* = 1,2,3,4,5-pentamethylcyclopentadienide). Higher yields (86%) and shorter reaction times were noted in the presence of an excess of KC8 (2 equiv.) than what were previously reported using sodium amalgam. Characterization of 2 by 1H NMR spectroscopy confirmed formation of the desired product, Cp*2U(2,2'-bpy) (2). Further characterization of 2 using IR spectroscopy showed sharp absorption bands at 915 cm-1 and 1499 cm-1, indicative of a reduced radical anionic bipyridine ligand.
Compound 2 reacts with a variety of carbonylated biomass mimics, including p-tolualdehyde, furfuraldehyde, acetone and benzophenone. Upon addition of one equivalent to 2, a color change from dark green to red-pink for aldehydes and to purple-pink for ketones ensues. The respective complexes, 3a-3d, were produced in excellent yields (87-94%) (eq. 1). Characterization by 1H NMR spectroscopy shows similar features for the whole family and supports Cs symmetry. Analysis of 3a by X-ray crystallography showed a bent metallocene framework with coordinated bipyridine and the oxygen of the carbonyl group. Comparison of the U-N and U-O bond lengths establishes a uranium(IV) species with a new carbon-carbon bond formed between the carbonyl carbon and the carbon of the bipyridine ring. Structural parameters of 3a show that the bipyridine ring is no longer reduced. Bond lengths of the bipyridine in 3a are typical for C-C single bonds, while angles support sp3 hybridized carbon centers. Further characterization of this family was accomplished by electronic absorption spectroscopy, which showed sharp, low intensity bands assignable to f-f transitions over the near-IR region, characteristic of f 2 uranium(IV) compounds.
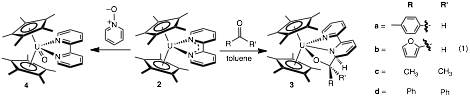
A possible mechanism for the formation of complexes 3a-d involves coordination of the substrate through the carbonyl oxygen, followed by reduction of the C=O bond by one electron from the uranium center, oxidizing it to U(IV). This radical can then couple to the radical delocalized over the reduced bipyridine ligand, forming complexes 3a-d. A similar mechanism has been previously noted for uranium(III) generated in situ.
The reactivity of 2 with oxygen atom transfer reagents was also explored for synthesis of terminal uranium oxo complexes. Treating 2 with an equivalent of pyridine-N-oxide affords the tetravalent uranium oxo, Cp*2U(2,2'-bpy)O (4), indicating that 2 can act as a source of divalent uranium (eq. 1). Characterization by 1H NMR spectroscopy and X-ray crystallography confirmed product formation. The molecular structure shows a bent metallocene framework with a terminal oxo ligand and a neutral bipyridine ligand. This species reacts with trimethylsilylhalides to cleanly generate the Cp*2UOSiMe3(X) (X = Cl, I). The reduction chemistry of 4 is currently underway to determine if trivalent uranium oxo species can be realized with this system.
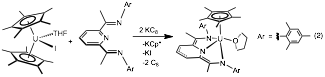
In addition to bipyridine, the redox active pyridine(diimine) ligand was explored. In analogy to 2, 1 was treated with 2 equivalents of KC8 in the presence of one equivalent of ligand. During the reaction, one Cp* dissociates, and the product is isolated as Cp*U(PDI)(THF) (4) (eq. 2). Characterization by 1H NMR and electronic absorption spectroscopies, as well as X-ray crystallography, all support the formulation as a uranium(IV) species. The bond distances of the pyridine(diimine) ligand in 5 are consistent with reduction by three electrons. This highly reduced uranium complex may act as a source of monovalent uranium, and the reactivity of this species is currently being explored for C-O bond cleavage.
Recently, we have begun exploring sterically bulky cyclopentadienyl ligands with a dimethylphenyl substituent (CpPH) to support reactive electron-rich uranium centers. Treating UI3(dioxane)1.5 in THF solution with either one or two equivalents of the new Cp ligand has afforded the metallocene species, CpPUI2 (6) CpP2UI (7). Characterization by 1H NMR spectroscopy supports formation of these new species, and efforts are underway to characterize both sterically bulky species by X-ray crystallography. Our focus over the remaining year will be to generate redox-active ligand complexes of 6 and 7 to test their reactivity for C-O bond activation as well as oxygen atom transfer.
