www.acsprf.org
Reports: UR349334-UR3: Photocatalytic Dechlorination of Chloroalkanes in Hydrocarbon Mixtures
The goal of this PRF-funded research is to develop catalytic methods to remove chloroalkanes impurities from petroleum by photochemical degradation. In our studies thus far we have used cyclohexane as a stand-in for petroleum and chloroform as the chloroalkane. We have followed the photodegradation process through the accumulation of decomposition products during irradiation.
The proposed work was based on two hypotheses. One was that metal complexes irradiated by visible or near UV light would be used to create radicals in solution, by any of several mechanisms. For example, using a chlorometallate complex (abbreviated MCln+), the following sequence can take place:
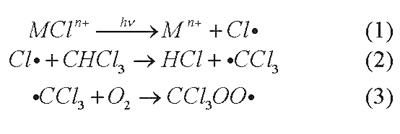
The trichloromethylperoxy radicals go on to produce phosgene, which can be converted to CO2 and HCl by exposure to water.
The second hypothesis was that the photodegradation would occur even at very low chloroform concentrations, because the decomposition would still take place even if hydrogen were abstracted from the hydrocarbon rather than chloroform itself. This is because the resulting alkyl (or aryl) radical would, when O2 is restricted, be able to abstract hydrogen only from CHCl3.
In the first few months of the project (the grant became active on July 1, 2009) we tested these hypotheses with (Bu4N)2FeCl4, which has high photocatalytic activity in pure chloroform, as a homogeneous catalyst. The rate of photodecomposition was measured in different ratios of chloroform to cyclohexane. What was observed was a small diminution in the degradation rate as the concentration of chloroform decreased. A solution with 20% chloroform still had a decomposition rate about 90% of the rate found in pure chloroform. Unfortunately, we could not carry the experiments down to lower concentrations, because the catalyst was insoluble. We expect this to be a common problem.
In September of 2009 the project was put on hold as I began a year-long sabbatical leave at the University of Ferrara, in Italy. I worked with Prof. Andrea Maldotti, whose area of specialty is heterogeneous catalysis by coordination complexes. When I returned my vision of the PRF project had changed, particularly in that the main goal was modified to develop a heterogeneous catalyst. This had been a long-term goal in the original proposal, but it became our sole focus this past year. This had the immediate advantage of eliminating the solubility problem in solutions with a high hydrocarbon percentage, since the catalysts were required to be insoluble in the first place.
Our objective was to examine catalysts that operated by several different mechanisms to produce radicals. Given the chloroform/cyclohexane model system, we intended to try catalysts for each of the following mechanistic pathways, categorized by what occurs to the excited state metal complex, represented for convenience as MCln+.
1. Photodissociation of chlorine atoms (as described above) to create Cl radicals.
2. Photooxidation to MCl(n+1)+, reducing chloroform to Clˉ and CHCl2 radicals.
3. Photoreduction to MCl(n-1)+ , oxidizing the counterion to a radical
4. Energy transfer to chloroform, which dissociates to Cl and CHCl2 radicals
We had previously found homogeneous catalysts that worked by the first three mechanisms. Energy transfer was considered a fourth possibility because, even though we had not seen any examples of photocatalysis by energy transfer, benzene is known to catalyze the photodecomposition of chloroform by this route.
Our plan was to use anionic chlorometallate complexes, immobilizing them on an anion exchange resin. By this means we found two metal complexes that appeared to catalyze chloroform decomposition. During control experiments, however, we discovered that the catalytic agent was not the metal complex, but the polystyrene anion exchange resin itself. Consequently, we investigated that process in detail, with the manuscript now nearing completion.
The relevant information for the PRF project is that, while the Dowex anion exchange resin (in the Clˉ form) catalyzes the photodecomposition of pure chloroform, admixing any cyclohexane quenches the reaction. Unlike the homogeneous photocatalysis we had earlier investigated, even 0.1% cyclohexane completely stopped photodegradation. Upon further investigation we discovered why. Rather than any of the mechanisms we had expected, based on results with homogeneous catalysts, a completely different mechanism was in play. Light was absorbed by the polystyrene chromophores (probably forming excimers), which reduced O2 to O2ˉ. The superoxide ion then attacked chloroform in the vicinity of Clˉ anions, yielding CCl4 initially, which decomposed further in a secondary reaction to phosgene. Cyclohexane quenched the reaction by associating closely with the styrene units, preventing access of O2 and CHCl3 to excited state polystyrene.
We next moved to mesoporous silica (MCM-41) as the support for chlorometallates. Again we found candidates that catalyzed the photodegradation of pure chloroform. However, again control experiments revealed that the photocatalytic activity was derived solely from the silica. This was a surprise, since ordinary silica had no catalytic activity whatever. We therefore began studying mesoporous silica as a photocatalyst on its own. Though the degradation products are similar to those found with the polystyrene anion exchange resin as the catalyst, the mechanism differs in important details. One experimental sign is that water quenches the photodegradation with mesoporous silica as the catalyst, but not when the polystyrene resin was the catalyst. The opposite is true with cyclohexane. It does not quench the photoreaction in the presence of mesoporous silica. Experiments are currently underway with increasing amounts of cyclohexane.
One thing we can say about the mesoporous silica-catalyzed photodegradation, based on product distributions, is that the mechanism of photocatalysis is not any of the four listed above. We are therefore moving ahead with experiments designed to foster one or another of the four mechanisms.