www.acsprf.org
Reports: AC148079-AC1: Asymmetric Halogen-Metal Exchange of Geminal Dihalides with Planar Chiral Organometallic Reagents
Introduction
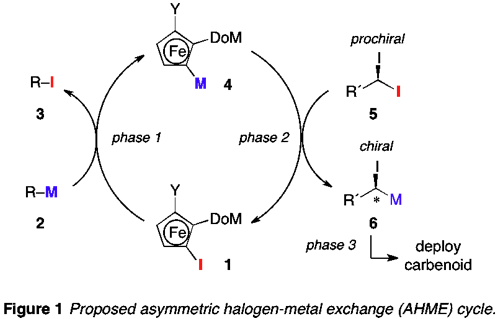
The goal of the funded research was to realize an asymmetric halogen-metal exchange (AHME) reaction of geminal dihalides using a recyclable planar chiral organometallic reagent derived from an iodoferrocene. The proposed AHME process (Figure 1) is a closed cycle in which a recyclable planar chiral ferrocene is used as a stereoinductive element. The complete AHME cycle incorporates three distinct phases: [1] generation of a planar chiral ferrocenylmetal (4) from an iodoferrocene precursor (1) via iodine-metal exchange using a simple achiral alkylmetal reagent (2); [2] AHME between 4 and a prochiral geminal diiodide (5) leading to the regeneration of 1 and the formation of a chiral a-haloalkylmetal (6); and [3] trapping of the scalemic carbenoid 6 in some process of interest (herein, reaction with aldehydes).
Results and discussion
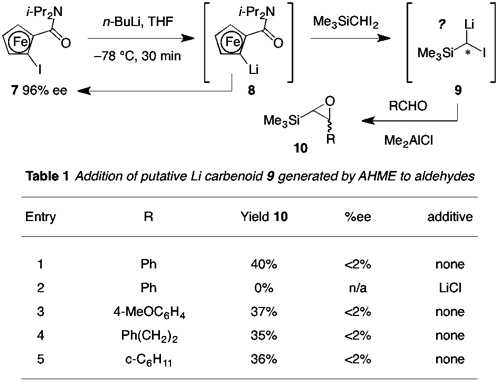
RECAP: In previous work (refer to earlier reports for details), optimum methods for the synthesis of planar chiral ferrocenyl iodides 1 (DoM = CONR2, SOAr, oxazolinyl) and geminal dihalide reagents (both diiodides 5 and chloroiodides, R' = alkyl, SiMe3) were developed. It was found that ferrocenes 1 with Y H are difficult (or else impossible) to prepare but that for easily accessed examples where Y = H, racemization of lithium reagent 4 does not occur (thus, one important reason for desiring Y H in the first place is eliminated). Validity of the complete AHME cycle was realized for the first time using benzaldehyde as the probe electrophile and TMS diiodomethane as the carbenoid precursor; precomplexation of the aldehyde with a Lewis acid (Me2AlCl) was critical for success in this endeavor.
SCOPE: After validating the basic premise of the AHME cycle in a limited context, studies shifted to establishing the scope of the aldehyde trapping variant of the process and determination of enantiomeric excess (%ee) for isolated products. Focusing initially on Li based chemistry, it was found that addition of putative (trimethylsilyl)iodomethyllithium (9) to a range of aryl and alkyl aldehydes gave Darzens-type epoxide products 10 in modest yield (e.g., Table 1). As expected, iodoferrocene 7 was recovered from the reaction without compromized enantiomeric excess; however, products 10 were found to be racemic.
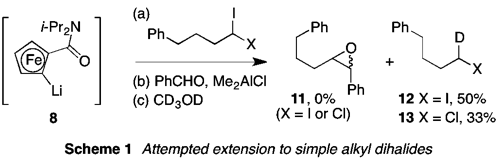
AHME using lithium 8 (generated as before) and Ph(CH2)3CHI2 or Ph(CH2)3CHClI was similarly disappointing (Scheme 1). In this case, putative carbenoid species failed to add to the aldehyde test electrophile at all, although it was possible to intercept them with deuterium. In ongoing experiments (vide infra), we are attempting to establish whether deuteration occurs with any level of stereocontrol.
Analogous reactions initiated by the Grignard based congener of 8 (generated from 8 by transmetallation with MgBr2) failed at their semination point. The magnesiated ferrocene did not participate in iodine-metal exchange reactions to any significant extent with any one of four geminal dihalide compounds: Me3SiCHI2, Me3SiCHClI, Ph(CH2)3CHI2, or Ph(CH2)3CHClI.
CONTROL EXPERIMENTS: It became apparent during the course of our studies that putative alpha-haloalkylmetal reagents 6 generated by addition of ferrocenyl metals 4 do not behave in the same way as ostensibly identical species formed by alternate routes, e.g., via deprotonation, or (more surprisingly) via iodine-metal exchange from common precursors using n-BuLi or EtMgCl. Thus, for example (Scheme 2), iodine-metal exchange from 14 (X = Cl or I) using n-BuLi followed by addition of PhCHO gave Darzen's product 11 in acceptable yield but, as noted above, the same compound was not formed when ferrocenyllithium 8 was used to initiate the same process. Two scenarios are possible: (1) ate-complex 15 proceeds to alpha-haloalkylmetal 16 (which is capable of adding to PhCHO) but ate-complex 17 does not (and cannot itself add effectively to PhCHO), or (2) alpha-haloalkylmetal 16 is not involved in either reaction and ate-complex 15 can add directly to PhCHO but ate-complex 17 cannot.

Conclusion and Impacts
In summary, after solving synthetic problems to access key reagents and substrates, and establishing that Y H was not required to maintain ferrocenyllithium stereochemical intergrity, we succeeded in validating a complete AHME cycle in a limited context. However, the process did not yield non-racemic products and it was found to have narrow scope. Control experiments suggest that nucleophilic transfer occurs from a persistent ferrocenyl iodine ate-complex rather than the putative alpha-haloalkylmetal reagent (6) itself. In the absence of SET during addition, stereoinduction is not precluded by such a mechanism. To test this hypothesis a few remaining experiments using deuterium as a probe hard electrophile await their conclusion in our laboratory. Whatever their outcome, it is evident that AHME as originally envisioned for the generation of main-group chiral carbenoids from geminal dihalides by the action of ferrocenylithiums is not a viable process. This fact does not, however, imply that the same kind of recyclable planar chiral organometal reagents could not be used to effect other types of useful processes that require the differentiation of enantiotopic iodine atoms.
Finally, the impact that this grant had on both the Blakemore Research Group (BRG) and the student conducting the research, Christopher Emerson, cannot be understated. Funding from the ACS PRF came at a critical juncture when the BRG was otherwise out of money, enabling us to continue our work and go on to secure additional support from the NSF. Also during the course of the grant, the PI was granted promotion with indefinite tenure at OSU and on the basis of his excellent research record, Christopher Emerson secured a post-doctoral position at Stanford University. Chris shall defend his PhD thesis on AHME (and other aspects of scalemic carbenoid chemistry) in November 2011 and will start his post-doc in January 2012.
DISSEMINATION: To date, research findings have been presented by Christopher Emerson at the 239th ACS National Meeting (3/21/10-3/25/10, San Francisco) and at the 1st Annual Graduate Research Symposium of the Organic Division of the ACS (7/15/10-7/18/10, Boston). More recently, Paul Blakemore presented the work at the Gordon Research Conference on Natural Products Chemistry (7/24/11-7/29/11, Smithfield, RI). Once all remaining studies are concluded (next 3-4 months), the results of the entire investigation will be compiled into a full paper for submission to the Journal of Organic Chemistry.