www.acsprf.org
Reports: ND649930-ND6: Energetics of the Ligand and Solvent Coordination of Catalytically Important Organometallic Complexes
Introduction
Using the newly constructed imaging Photoelectron Photoion Coincidence (iPEPICO) experiment of the new VUV beamline of the Swiss Light Source synchrotron, we have embarked on a project to determine highly accurate bond energies in the gas phase, especially of metal-ligand bonds. In the first phase of the experiments, we have perfected the experimental procedure and the data analysis and have published a number of papers on the PEPICO experiments of smaller molecules as well as organometallics and have published the PEPICO data analysis software.
Small molecule studies related to combustion thermochemistry
The dissociative photoionization onsets of Br and I loss reactions were measured for C2H5Br and C2H5I by PEPICO in order to establish the heats of formation of these ethyl halides and the ethyl cation that interconnects them. In establishing the thermochemistry of EtBr and EtI, a more accurate ethyl cation heat of formation was published, based on a recent value for ethyl radical heat of formation and on ab initio isodesmic calculations using recent PEPICO data. With this anchor, accurate heats of formation were obtained for EtI and EtBr, which are more consistent with the higher EtBr literature values, contrary to recent findings. For EtI, the latest calorimetric value does not agree within the claimed accuracy.
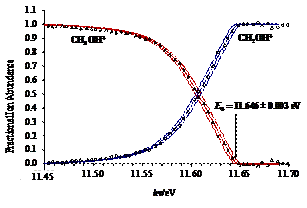
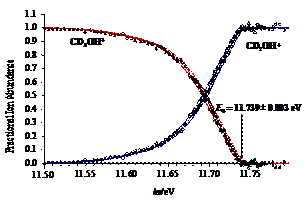
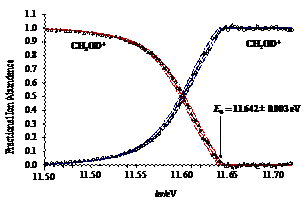
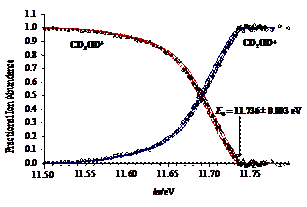
The dissociative photoionization of energy selected methanol isotopologue (deuterated) cations was investigated using imaging PEPICO. The first dissociation is an H/D-atom loss, while at higher energy, a parallel H2-loss channel weakly asserts itself. At photon energies above 15 eV, in a consecutive hydrogen molecule loss to the first H-atom loss, the formation of CHO+/CDO+ dominates, with little evidence for H-atom scrambling. In the photon energy range corresponding to the B˜ and C˜ ion states, a hydroxyl radical loss appears which process is confirmed to take place on the first electronically excited state. From the four independent appearance energies of the isotopologue ions and the calculated ZPEs, accurate heat of formation was determined for CH2OH+. This value then yields the heat of formation of one of the most fundamental species in combustion processes, the CH2OH radical.
Organometallics
Three metallocene ions were studied by PEPICO to investigate the mechanism, energetics and kinetics of their ionic dissociation processes. The energy-selected ions fragment by losing cyclopentadienyl ligand, while CH and C2H2 losses appear as minor channels. A possible isomerization pathway has been observed for Cp2Ni+, yielding a complex with pentafulvalene. The data was modeled by RRKM and the simplified statistical adiabatic channel model. Using SSACM, which seems more reliable for loose transition states, appearance energies and, with the ionization energies, ionic bond energies were determined for the cyclopentadienyl-loss dissociations. DFT calculations were used to determine the structures of these complexes and to help the analysis of the experimental data. The trends in the M-Cp bond energies were related to the electronic structures of the metallocene ions based on simple MO theory.
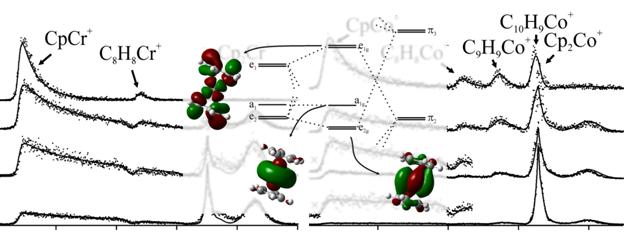
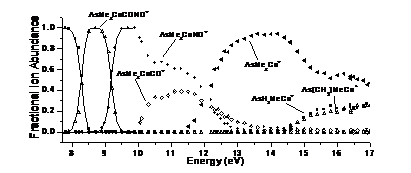
PEPICO spectroscopy was used to study a series of cobalt-organic complexes with phosphine and phosphine-analog ligands. The first two dissociations were carbonyl losses. Nitrosyl loss is a minor channel from the molecular ion and a major competitive channel from the first daughter ion. Further sequential CO/NO losses lead to the ER3Co+ ions, which, similarly to an earlier TCID study, exhibited ethylene loss and methane-loss dissociations. RRKM calculations to model the carbonyl losses yielded Co-CO bond energies, which were related to the electron donor and acceptor properties of the phosphine analog ligands. While earlier we have shown that uncomplexed EMe3+ ions (E: P to Bi) preferably lose a methyl radical, the cobalt-complex experiments showed that the transition metal acts as a catalyst to promote both carbon-carbon and carbon-hydrogen bond formation leading to either ethylene or methane loss from the complexed ions.
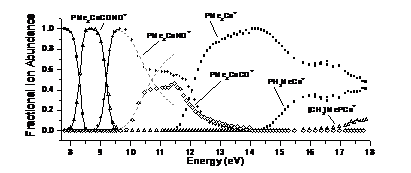 |
 |
 |
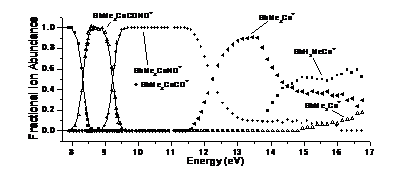 |
Recently,
at the SLS iPEPICO, using the molecular beam inlet source, we were able to form
adducts of an organocobalt complex with alkanes and recorded coincidence
spectra for the first time. The first successful experiments yielded hexane and
propane adducts, where we were able to produce both C3H8.CpCo(CO)2+
and C6H12.CpCo(CO)2+.
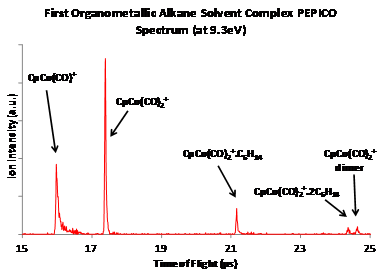
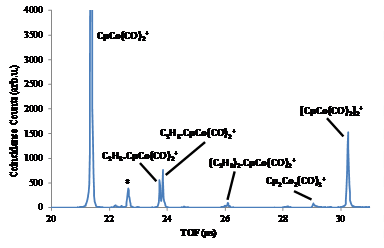
A
side product of these experiments was a very interesting dimer of the parent
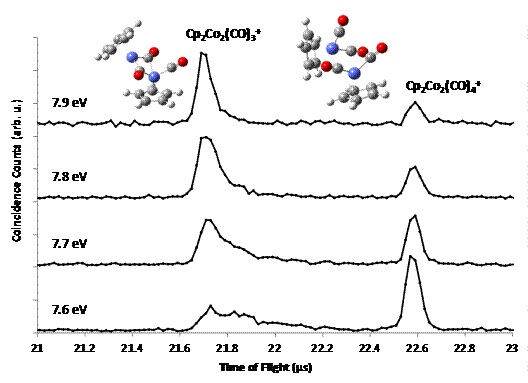
cobalt complex, Cp2Co2(CO)4 that ionized to Cp2Co2(CO)4+.
This ion has only been formed by electrochemical oxidation before and has a
unique structure with no bridging carbonyls and a weak cobalt-cobalt bond. In
our experiments, we observed this cation losing a carbonyl ligand and forming a
more stable dinuclear complex. This tricarbonyl complex has been isolated in
the neutral form before and is either triply or singly bridged while our
calculations indicate that the ion has a doubly bridged structure. In our
latest experiments, we were able to form the precursor dimer ion and measure
the absolute dissociation kinetics into the tricarbonyl dimer cation showing that
the tricarbonyl complex is not formed as a neutral, later ionized by the VUV
radiation, rather the Cp2Co2(CO)4+
ion loses a carbonyl ligand to form the bridged tricarbonyl dimer ion.
Data Analysis
A computer program has been developed to model and analyze the data from PEPICO experiments, which models the dissociative photoionization process by calculating the thermal energy distribution of the neutral molecule, the energy distribution of the molecular ion as a function of the photon energy and the resolution of the experiment. Parallel or consecutive dissociation paths of the molecular ion and also of the resulting fragment ions are modeled to reproduce the experimental breakdown curves and time-of-flight distributions. The internal energy distribution of the fragment ions is calculated from the densities of states using the microcanonical formalism to describe consecutive dissociations. Dissociation rates can be calculated by the RRKM, SSACM, or VTST rate theories, and can include tunneling effects. Isomerization of the dissociating ions can be considered either from the original or the isomer ions. The program, which can optimize the various input parameters to find a good fit to the experimental data, is now made available to other users of the SLS iPEPICO apparatus.