www.acsprf.org
Reports: UNI550385-UNI5: The Role of Defects in Sulfur Removal Over Titanium Dioxide
Fundamental studies of the interaction of organosulfur compounds with oxides are important in understanding the role that oxides (and their defects) play in desulfurization. However, very few fundamental studies have been performed on such compounds, particularly the larger refractory species. Specifically, oxides act as :(1) supports for metal sulfide catalysts used in petroleum hydrotreaters to produce H2S from organosulfur compounds; and (2) catalysts in the Claus process to form elemental sulfur from H2S. To this end, we currently employ temperature programmed reaction spectroscopy (TPRS), Auger electron spectroscopy (AES), and X-ray photoelectron spectroscopy (XPS) to investigate the interaction of several sulfur-containing molecules (Figure 1) with a rutile titanium dioxide (110) surface.
Figure 1. Investigated species: benzenethiol, thioanisole, thiophene, tetrahydrothiophene
The above molecules, with the exception of benzenethiol which was studied for comparison, are present as core linkages in petroleum. If the sulfur from these core species is not fully removed, sulfur oxides formed during combustion can react with water in the atmosphere and lead to acid rain.
The TPRS data collected for benzenethiol and thioanisole are very similar, the latter of which is shown in Figure 2. Two main desorption features can be observed following exposure of the TiO2(110) substrate to thioanisole at 90 K. The highest temperature state appears at 395K for a surface coverage of 0.1 ML. This temperature corresponds to a desorption activation energy of approximately 105 kJ/mol assuming that the desorption is first order, and using a prefactor of 1013 s-1. Upon increasing the surface coverage to 0.4 ML, a broadening on the low temperature side and a shift of this state to 360K occurs, indicative of interactions between thioanisole molecules which lead to destabilized thioanisole-titania binding. Multilayer desorption is observed as a sharp peak at 200K which does not saturate with increasing coverage.
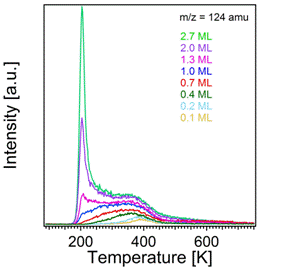
Figure 2. Temperature programmed desorption spectra of thioanisole from TiO2(110).
The TPRS traces for thiophene and tetrahydrothiophene appear quite different from one another, as shown in Figures 3 and 4, respectively. Thiophene desorption occurs in three main states. The highest energy desorption state, α1, occurs at 255K for a coverage of 0.1 ML, and this state shifts to 240K with increasing coverage while broadening on the low temperature side of the peak. A second desorption state, α2, develops at 170K, and finally the multilayer peak appears at 140K and does not saturate with increasing exposure. The α1 peak likely corresponds to adsorption to Lewis acidic surface Ti4+ sites via electron donation from the lone pair electrons on S. By analogy to behavior of thiophene on a metallic substrate (specifically Cu(111)),1 the α2 desorption state likely corresponds to a tilting upward of adsorbed thiophene once a critical coverage is reached and the adsorbed molecules start to interact with one another. Tetrahydrothiophene, however, shows only 2 main desorption states: one at 380K, α1, which saturates, and one at 160K which does not. A very weak, nearly indistinguishable amount of desorption is visible between 150K and 200K. Interestingly, the α1 state of tetrahydrothiophene is over 100K above that of thiophene. This stronger bonding can likely be attributed to the greater availability of the lone pair electrons on S in the absence of the aromatic ring of thiophene.
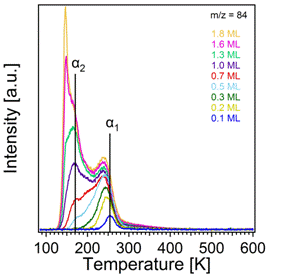
Figure 3. Temperature programmed desorption spectra of thiophene from TiO2(110).
For all molecules examined, a full scan over our mass spectrometer's 300 amu range revealed no additional desorption species attributable to reaction with the substrate, indicating that desulfurization over the vacuum annealed (reduced) TiO2(110) surface did not occur. We also looked at a heavily reduced TiO2(110) surface which was prepared using 1.5kV Ar+ ion bombardent. Such a surface contains significant amounts of Ti2+ and Ti3+ species in addition to Ti4+. On this surface, a fraction of the benzenethiol underwent reaction, producing benzene (data not shown) in the TPRS studies. This is likely the result of the binding of S to reduced Ti species on the surface. We are in the process of confirming this hypothesis by employing XPS to examine any remaining surface species. None of the other molecules produced evidence of reaction over a bombarded surface, indicating the enhanced stability of the organosulfur compounds (R2S) as compared to the thiol (RSH).
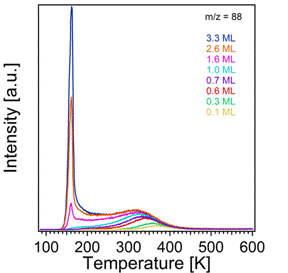
Figure 4. Temperature programmed desorption spectra of tetrahydrothiophene from TiO2(110).
We plan to examine more complex thiophenes, including benzothiophene and dibenzothiophene in preparation for investigating the desulfurization of these molecules by modifying the surface. These thiophenes are considered refractory as they are not fully removed following traditional hydrodesulfurization methods. In order to introduce these heavier, relatively low vapor pressure species we will need to modify our vacuum chamber to include a solid dosing system. This set-up is currently being built. We also purchased an Auger electron / X-ray photoelectron spectrometer (AES/XPS) with the funds provided from this grant, and have been routinely using AES to monitor the surface. XPS data collection is also underway.
These funds have provided fruitful research experiences for five undergraduate students so far, 2 of which had fulltime 10-week summer positions in addition to research time during the academic year. All students involved have had the unique opportunity to participate in instrument development as well as the collection and analysis of data. We plan to publish and present results from this work in the coming year.
Acknowledgment is made to the Donors of the American Chemical Society Petroleum Research Fund for support (or partial support) of this research.

1. Milligan, P. K.; Murphy, B.; Lennon, D.; Cowie, B. C. C.; Kadodwala, M., A complete structural study of the coverage dependence of the bonding of thiophene on Cu(111). Journal of Physical Chemistry B 2001, 105 (1), 140-148.