www.acsprf.org
Reports: UNI150347-UNI1: Toward Greater Understanding and Expanded Utility of the Palladium-Catalyzed Activation of Carbon-Carbon Single Bonds
The activation and functionalization of carbon-carbon single bonds remains an unrealized facet of organic chemistry. A generalized transition metal-catalyzed methodology allowing for the selective cleavage of carbon-carbon bonds has nearly boundless potential, yet efforts to date have primarily relied upon specialized substrates for the achievement of this unusual reaction.[1]
As with the methodology as a whole, there are very few examples of mechanistic investigations into transition metal-catalyzed carbon-carbon bond activation processes. To remedy this dearth of information, our group has initiated the mechanistic investigation of the palladium-catalyzed β-aryl elimination of triarylmethanols (Scheme 1), a reaction first reported by Nomura and coworkers.[2] Our intent is to uncover factors that influence the carbon-carbon bond step of this process, and then subsequently utilize this information for the extension of known reactivity.
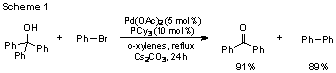
At the onset of the project, we were contemplating two general mechanistic hypotheses, differing primarily in the sequence of elementary steps of catalysis. In the first mechanism (Scheme 2), a Pd0 species oxidatively adds to bromobenzene to form organometallic species A. After ligand exchange, β-aryl elimination generates diaryl palladium compound B, which subsequently undergoes reductive elimination to produce benzophene and biphenyl. A second hypothetic mechanism (Scheme 3) includes similar fundamental steps, but begins with β-aryl elimination before proceeding via oxidative addition. This mechanism can be envisioned to proceed through either a Pd(0)/Pd(II) or Pd(II)/Pd(IV) redox cycle.

Our efforts to discern between these mechanistic hypotheses with two simultaneous approaches. Two students (one supported by a Moissan Undergraduate Fellowship sponsored by the ACS Division of Fluorine Chemistry) were involved in preparing a number of substituted triarylmethanols for the identification of factors, if any, that influenced the relative propensity of β-aryl elimination. A third student performed reactions of a more mechanistic nature to examine the reversibility of various steps throughout the catalytic reaction.
Approximately 30 aryldiphenylmethanol compounds containing variable substitution were prepared and subjected to the standard reaction conditions (Scheme 4). As indicated, several products are possible in these reactions, and the relative ratio of these products was determined utilizing GC/MS methods thus providing the means to examine the cleavage propensity of each substituent.
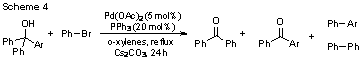
The influence of sterics upon β-aryl elimination is readily apparent. Aryl groups with o-substitution of any kind, even as sterically unimpressive as fluorine, demonstrated the a dramatic propensity to cleave selectively versus unsubstituted phenyl groups (Table 1). It is notable that the fluorine substituted compound demonstrated a propensity to cleave that extended far beyond its steric nature. It has been hypothesized that a potential metal interaction with the C-F bond influences this transfer (vide infra).
Table 1. Relative aryl group transfer rates | |
Ar | Ar:Ph |
o-Me-C6H4 | 15:1 |
o-F-C6H4 | 75:1 |
o-OMe-C6H4 | 20:1 |
Notably, variation of the electronics of an aryl group had only subtle influence on selectivity (Table 2). A series of substituents, varying from electron deficient 3,5-bis(trifluoromethyl)phenyl to electron rich 4-dimethylaminophenyl, demonstrated no clear trends—in fact, both more electron deficient and electron rich substituents appeared to increase the rate of β-aryl elimination versus the phenyl substituent, with the electron deficient substituents showing a more consistent effect.
Table 2. Relative aryl group transfer rates | |
Ar | Ar:Ph |
p-NMe2-C6H4 | 1.24:1 |
p-OMe-C6H4 | 1.23:1 |
p-Me-C6H4 | 1.4:1 |
m-Me-C6H4 | 1:1 |
p-F-C6H4 | 2.5:1 |
p-CF3-C6H4 | 3:1 |
3,5-(CF3)2-C6H3 | 5:1 |
A clearer picture was obtained with the preparation and testing of a series of fluorine substituted substrates, chosen with the hope of minimizing coordination effects and other variables potentially complicating the electronic effects. Results from this series of experiments is provided in Table 3. The significant influence of an ortho-fluorine is readily apparent, and brings to mind studies implicating interactions of metal centers with C-F bonds in stabilizing organometallic intermediates.[3] Beyond the ortho-substitution, additional electron withdrawing substituents result in greater selectivity, and the inclusion of multiple fluorine atoms results in cumulative effects.
Table 3. Relative fluorinated aryl group transfer rates | |
Ar | Ar:Ph |
4-F-C6H4 | 2.5:1 |
3-F-C6H4 | 4:1 |
2-F-C6H4 | 76:1 |
3,4-F2-C6H3 | 8:1 |
2,5-F2-C6H3 | 190:1 |
2,6-F2-C6H3 | 240:1 |
3,5-F2-C6H3 | 11:1 |
3,4,5-F3-C6H2 | 50:1 |
Additional mechanistic studies focused on the analysis of steps within the proposed catalytic cycle. A series of experiments were designed to examine each step in turn and to ascertain the influence of each component upon the overall reaction.
Initial efforts focused upon changing the aryl bromide used for coupling. This was performed by simultaneously utilizing two aryl bromides, stopping the reaction short of complete conversion, and then determining the ratio of the products formed by GC/MS or NMR methods. Results from a series of these competition reactions demonstrated that electron deficient aryl bromides, such as those containing trifluoromethyl substitution, proceeded more rapidly than electron rich aryl bromides (Scheme 5). In summary, this indicates that oxidative addition either limits catalyst turnover or lies in a reversible step that precedes the turnover limiting step.
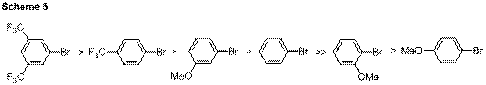
The reversibility of the carbon-carbon bond activation step was examined by running the reaction under standard reaction conditions in the presence of an additional ketone. In this case, the desired products, benzophenone and biphenyl, were observed, but no sign of mixed triarylmethanol or benzophenone species were observed, indicating that β-aryl elimination is either a reversible process or the resulting intermediate is not sufficiently stable to allow ketone exchange prior to further reaction.

To date, this work has provided significant insight into factors influencing β-aryl elimination, including the effects of electronics on the C-C activation step as well as insight into the catalytic cycle. Ongoing efforts will further this kinetic study and promise to clarify the mechanism of this process. The work will continue with the expansion of this known methodology toward more widely applicable coupling methodologies utilizing carbon-carbon bond activation as an entry point into high energy organometallic species.