ACS PRF | ACS
All e-Annual Reports

44683-G1
Guanidinium Catalysts for the Asymmetric Addition of Nuecleophiles to N-Acylhydrazones
The original project was the development of reactions promoted by guanidinium catalysts. These hydrogen bond catalysts would activate N-acylhydrazone electrophiles for attack by nucleophiles. Initial studies on this project were unsatisfactory. Our focus then turned towards a project that was showing promise. This project involved the kinetic resolution of secondary alcohols which is an important transformation for the preparation of chiral building blocks. While there are a number of ways to selectively derivatize one alcohol enantiomer over another, the use of a silyl group has been virtually ignored. Therefore we are working on the asymmetric silylation of secondary alcohols via two different approaches: chiral cation catalysts (Eq. 1) and chiral nucleophilic catalysts (Eq. 2). Both approaches have shown promising preliminary results in achieving enantioselectivity.

Chiral Cation Silylations
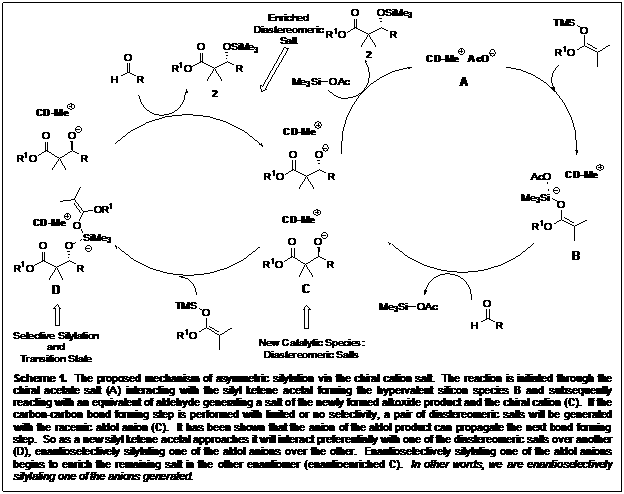
In the process of investigating a new, asymmetric version of the Mukaiyama aldol reaction we discovered something new. We determined that the enantioselectivity of the two products (the alcohol and silylated product) arose through enantioselectively silylating the racemic alcohol (Scheme 1), not through the carbon-carbon bond forming step. Recognizing the value of an enantioselective silylation reaction, we proceeded to target this aspect of the reaction.
Scheme 1. General schematic depicting the proposed reaction pathway of (A) the chiral cation mediated reaction and (B) the chiral nucleophile activated pathway.


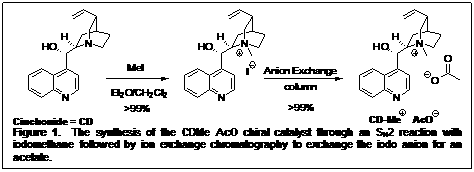
The design of the system incorporates the generation of a chiral cation salt between a secondary alkoxide anion and a chiral ammonium cation to facilitate the selective silylation of one of the alkoxides. The alkoxide is generated in situ and results from the reaction between a silyl ketene acetal and an aldehyde through activation by a Lewis base (an acetate) (Table 1). Cinchona alkaloids were chosen as the counter ion (Scheme 1). The catalysts are easily synthesized through nucleophilic substitution on the quinuclidine core. Figure 1 shows the synthesis of the methyl derivative of cinchonidine.
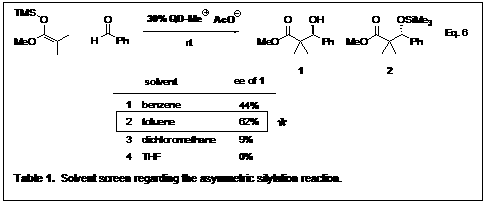
The quinidine methyl acetate (QDMeAcO) catalyst was the first catalyst synthesized to screen reactions. Benzaldehyde and methyl trimethylsilyl dimethylketene acetal were the substrates chosen for the screening. Solvent was one of the first parameters investigated. Table 1 shows four of the solvents investigated, showing toluene was the best solvent. The proposed mechanism of our enantioselective silylation is shown in Scheme 2. Employing a chiral cation acetate salt in the enantioselective silylation reaction allows for the direct formation and silylation of an alcohol in one pot starting from a silyl ketene acetal and an aldehyde.
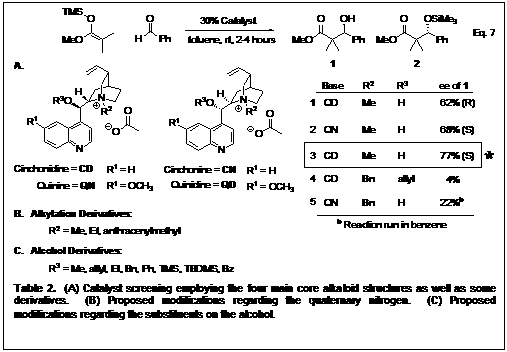
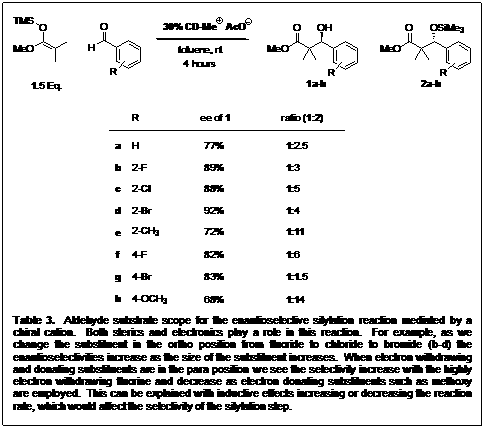
Since we are employing a cinchona alkaloid as the chiral cation for our system, we have four different core structures available for testing. The four compounds are available in two pairs of diastereomers (Table 2). We therefore proceeded to derivatize them for investigation in our reaction. Indeed, the QN/QD diastereomer pair acted as psuedoenantiomers (Table 2) but the cinchonidine structure gave higher enantioselectivity overall. We also screened other functionalized catalysts, but they did not perform as well as CDMeAcO. Next we looked at the substrate scope with regards to the aldehyde, incorporating a variety of sterics and electronics. Table 3 shows the enantioselectivities of 1a-h obtained from the reaction of methyl trimethylsilyl dimethylketene acetal and different aldehydes, showing that sterics and electronics play a role.
Nucleophilic Enantioselective Silylations.
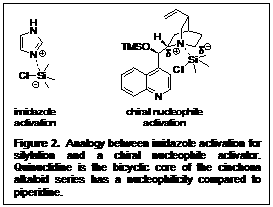
While our initial asymmetric silylation reaction was quite novel, it was complex, therefore we wanted to develop a system that was more general. So we began thinking about the mechanism of silylating an alcohol. It has been postulated that the first step in silylating an alcohol is a pre-equilibrium between the silylating reagent and a nucleophile activator. Therefore, we reasoned that a chiral nucleophile could be used to selectively silylate one alcohol enantiomer over another (Figure 2). A series of catalysts were synthesized to protect the secondary alcohol. For the first round of screening, the derivatizations included: trimethylsilyl, methyl, ethyl, and benzyl (Figure 3).

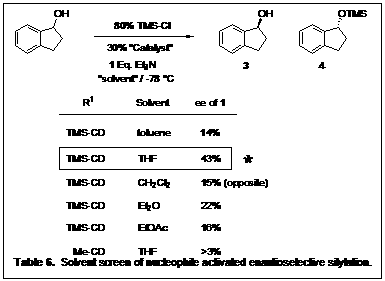
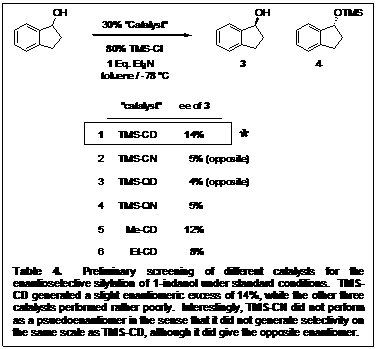
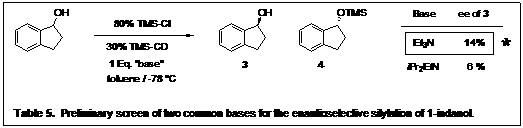
To test the catalyst substructure, all four of the alkaloids were derivatized with the trimethylsilyl group and 1-indanol was the secondary alcohol chosen for the screening. Initial reactions were screened in toluene with trimethylsilyl chloride, and triethylamine. Table 4 shows TMSCD to be the optimal catalyst.
Initial reaction screenings were performed with triethylamine as the base for neutralization of the HCl generated. As an initial screen we have also looked at Hünig's base (Table 5). Surprising, triethylamine still had higher enantioselectivity. We intend to look at other bases as we continue to optimize our conditions.
With regards to solvent, we initially screened a few to investigate their affect on the reaction. Since our reactions are run at low temperatures, we were limited to solvents that did not freeze at -78 °C. The reactions were screened using the TMSCD catalyst (Table 6) and THF was shown to achieve superior selectivity over other solvents tested.