ACS PRF | ACS
All e-Annual Reports

44477-G6
Using Chemical Energy at a Single Molecule Level: Chemomechanical Coupling of Molecular Motors
During the last year, our research has been focused on the understanding of the molecular mechanisms of chemical energy usage by motor proteins and the development of molecular dynamics simulation methods of studying slow conformation changes. We focused on the modeling of the kinetics of kinesin walking in the presence of external load and development of simulation methods for the atomic detailed simulations of protein motions.
1. Chemo-mechanical simulations of motor proteins
The hand-over-hand mechanism of kinesin walking
Earlier, we proposed a theoretical model for the understanding of the experimental observations on the force dependence on the walking velocity and forward/backward ratio for kinesin. The model reproduces the hand-over-hand mechanism for kinesin walking to the plus-end of microtubule. The model predicts that both forward and backward motions of kinesin take place at the same chemical state of the motor heads, with the front head being occupied by an ATP(or ADP,Pi) and the rear by an ADP. The direction of motion is a result of the competition between the power stroke produced by the front head and the external load. The model predicts, in agreement with experiments, that the chemomechanical coupling remains tight in a large force range. However, when the external force is very large (e.g., ~18 pN), kinesin slides in an inchworm fashion and the translocation of kinesin becomes loosely coupled to ATP turnovers.
In the continuation of our studies on the walking mechanism of kinesin, we studied the asymmetry in its walking. Experiments have shown that although the conventional kinesin 1 is a homodimer, it walks by an asymmetric hand-over-hand mechanism. We showed that the asymmetry in the walking mechanism, as suggested by others, can be a result of the different lengths of the neck-linker when different kinesin head takes the leading position. We also studied the experimentally suggested processive motion of man-made heterodimers of kinesin, in which only one of the two heads has the capability of ATP hydrolysis. We showed that the processive walking of these artificially constructed kinesins can be explained by involving a force generating intermediate, the kinesin-ATP complex. The model was then applied to study the force dependence of the “limping factor” of various kinesin construct, as well as on the walking of kinesin in the presence of both ATP and AMPPNP. Good agreement was obtained between the theory and experiments.
2. Development of selective transition path sampling method for accelerated rate constant calculations.
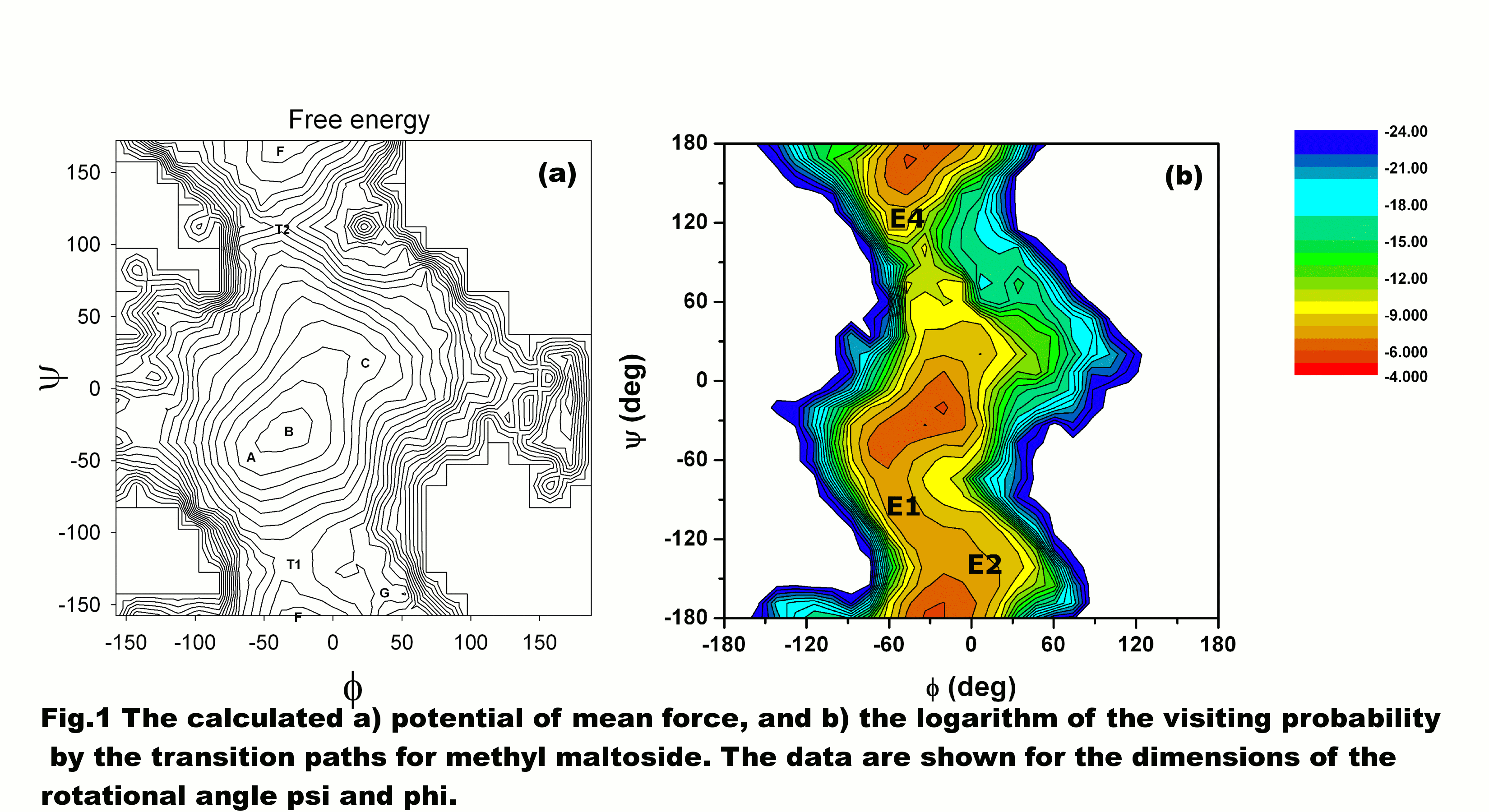
We developed a new and simple trajectory shooting method for the sampling of transition paths and for the calculation of rate constants. The method makes use of the accelerated MD simulations that allow much enhanced sampling of the “reactive” configuration space and to separate the reactive phase space from the non-reactive. The accelerated MD simulation is used to generate the initial phase space points for a selective sampling of successful transition paths over high energy barriers. The method accelerates the transition path sampling and rate constant calculations by significantly reducing the sampling of non-reactive trajectories, and at the same time calculates the ratio between reactive and non-reactive transition paths quickly. Further, as a result of these simulations, both thermodynamic (see Fig. 1a for a potential energy surface for the conformational change of methyl maltoside calculated using this method) and dynamical information can be easily obtained for the original system at all desired temperatures (as long as the distribution of sampled energy covers the energy distribution at the given temperature). This method also conserves in part the “throwing rope in the dark” advantage of TPS in that it does not require the knowledge on the reaction coordinate and thus is potentially useful for systems without known reaction coordinates and for those with multiple reaction paths. The method is expected to be useful in identifying new reaction pathways. The main difference between the present method and the standard TPS is that instead of generating transition paths from a pre-selected reactive one, the shooting of trajectories was initiated from an ensemble of phase space points. The sampling over reactive transition paths is enhanced through the enhanced sampling of initial configurations that more likely lead to reactive paths. The enhanced sampling of the more reactive region reduces the computational requirement of the large less reactive phase space and provides a quick estimate of the rate constant, although it might slows down the convergence in the rate constant calculation, due to the neglect of these less reactive points (not the rather large error bar of the calculated rate constant). The method was applied to study the conformational change of methyl maltoside. This study not only allows a fast calculation of the rate constant but also provides the probability of the transition paths for the reaction, which is shown in Fig. 1b in comparison with the potential of mean force.