ACS PRF | ACS
All e-Annual Reports

44534-B3
Sites of Protonation and Ligand Migration in Bimetallic Organometallic Complexes
This report focuses on a computational chemistry study of some bimetallic organometallic complexes. Our computational modeling is closely connected to the experimental work performed in the laboratories of Dr. M. Cowie at the University of Alberta, Edmonton.
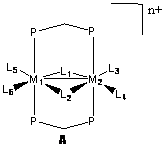
The purpose of this work is to examine fundamental catalytic conversions that occur at the bimetallic centers. The general structure is shown below in A, where the metal centers are from groups 8 and 9 in the periodic table. Though not involved in the chemistry in our study, the diphenyldiphosphinomethane ligand (dppm) provides the structural support necessary to hold the two metals in place. Around the metals are ligands occupying positions L1-L6, involved more directly in this study. Not all of these positions are necessarily occupied at all times, with positions varying based on the system being studied. The ligands involved include acetyl, carbonyl, hydride, methyl, methylene, phosphine, and triflate anion. The overall charge of the complexes includes neutral, cationic, and dicationic, depending on the ligands and metals involved.
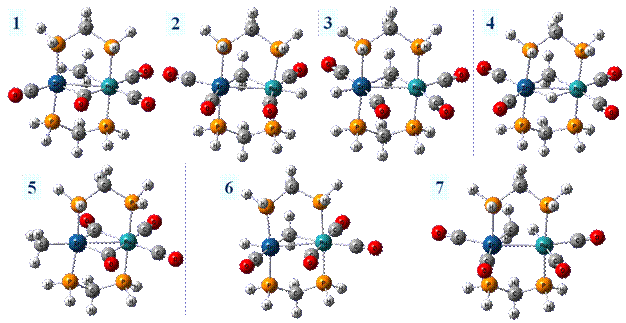
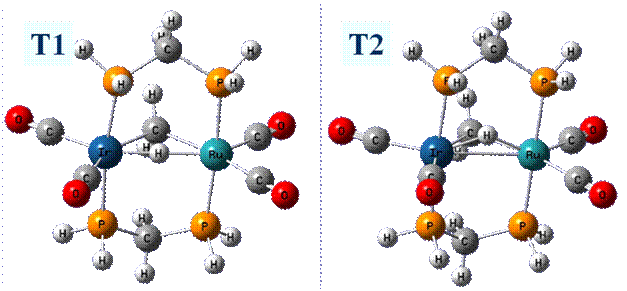
Gaussian03 was used for all calculations. The B3LYP theoretical method was employed for all calculations. The cc-pVDZ basis set was utilized for all atoms except the transition metal atoms, for which the LANL2DZ basis set was employed. The dihydrogen diphosphine methane ligand (dhpm) was utilized in place of the dppm ligand for our modeling work. We have examined seven sites of protonation and a hydrogen atom migration mechanism on [RuIr(CO)4(μ-CH2)(dhpm)2]+. Hence, the protonated molecule is a dication. The seven structures are depicted here. The most stable protonation structure is 4, which is a µ-H structure and corresponds to a structure observed by Cowie, et al. The energies of the remainder of the structures will be reported relative to this structure. The second site of protonation was on the bridging methylene, 1, from the iridium side. This resulted in an agostic bond of the methyl ligand to the iridium atom, with an energy of 3.3 kcal/mol. Next, protonation was attempted between the carbonyls on both metals. This led to the protonated Ir complex, 3, having a slightly lower energy, 13.7 kcal/mol versus 16.1 kcal/mol found for the protonated Ru complex, 2. We also found another protonated site on the iridium atom, 6, with a relative energy of 6.7 kcal/mol. Structure 3 has a carbonyl group situated between the µ-CH2 ligand and the terminal hydride ligand, whereas 6 has this carbonyl and terminal hydride interchanged. Structure 7, with an energy of 1.1 kcal/mol, has the protonation site such that a methyl group is agostically bonded to the Ru atom. Finally, 5 has a terminal methyl group on the iridium atom, with a relative energy of 0.1 kcal/mol. This work shows is that there are a number of low energy isomers that are possible in the protonation of these bimetallic systems. The Cowie experimental work showed that 5 could be formed as a low temperature transformation, starting from 4. These two structures are nearly iso-energetic. We next examined the migration pathway of the hydrogen atom from its bridging position between the metal atoms, to form the agostic methyl ligand group in form 1, which could subsequently rearrange to 5. Most depictions in the experimental literature show the hydrogen atom passing directly between the two metal atoms, to come out the “rear” side and form the agostic methyl ligand. Initial attempts to find the transition state resulted in structure T1 (see below). Compared to the hydrogen bridging position, the relative energy was 37.3 kcal/mol. Two imaginary frequencies were found, demonstrating that the structure is a second order saddle point, rather than a transition state. In an attempt to eliminate one of the imaginary frequencies, the hydrogen atom was held in the Ru-Ir-dhpm plane by freezing the two dihedrals from the two phosphorus atoms to the two metals to the hydrogen atom. This was allowed to optimize under that constraint to T2. While optimizing, the hydrogen migrated up in the Ru-Ir-dhpm plane. At the end of the optimization, the Ru-Ir-H bond angle was 22.2ş The energy was slightly lower than the T1 structure, having a relative energy of 36.2 kcal/mol. Only one imaginary frequency was obtained, indicating it was a transition state. We further relaxed the constraint on the Ru-Ir-dhpm plane, and this lowered the barrier height to 34.4 kcal/mol. All of these barrier heights are too high to explain the fact that the hydrogen atom migrates from the bridging position to the methylene group under low temperature NMR conditions. Hence, there must be another migration pathway for this mechanism. We have begun to explore such pathways.