
ACS PRF | ACS
All e-Annual Reports

40320-B4
[1,3] Sigmatropic Rearrangements in a Series of Bicyclo[4.2.0]oct-2-enes
The vinylcyclobutane-to-cyclohexene rearrangement has been known for over 40 years. This isomerization was formally classified as a [1,3] sigmatropic rearrangement in the The Conservation of Orbital Symmetry,1 and selection rules for [1,3] carbon migrations were enunciated: the si (suprafacial, inversion) and ar (antarafacial, retention) products were privileged as symmetry-allowed for monocyclic precursors; bicyclic vinylcyclobutanes, geometrically unable to form ar products, would have but one symmetry-allowed option, the si path.
The critical stereochemical and mechanistic issues pertaining to the vinylcyclobutane-to-cyclohexene rearrangement hinge on the question of concertness. Given that the energetics for this rearrangement are consistent with a stepwise nonconcerted reaction, degrees of stereoselectivity have been used as surrogate probes for concertness. Thus, the ratio of the rate of formation of the symmetry-allowed si product to that of the symmetry-forbidden sr product (the si/sr value) has become the default standard for assessing the degree of concert for [1,3] carbon shifts shown by bicyclic vinylcyclobutanes.While the mixed stereochemical results exhibited by the bicyclo[3.2.0]hept-2-enes2 have met with various interpretations, recent experimental and theoretical investigations have suggested that these reactions are almost certainly mediated by short-lived, nonstatistical diradical intermediates that partition to give multiple products from a common shallow plateau on the potential energy surface.3
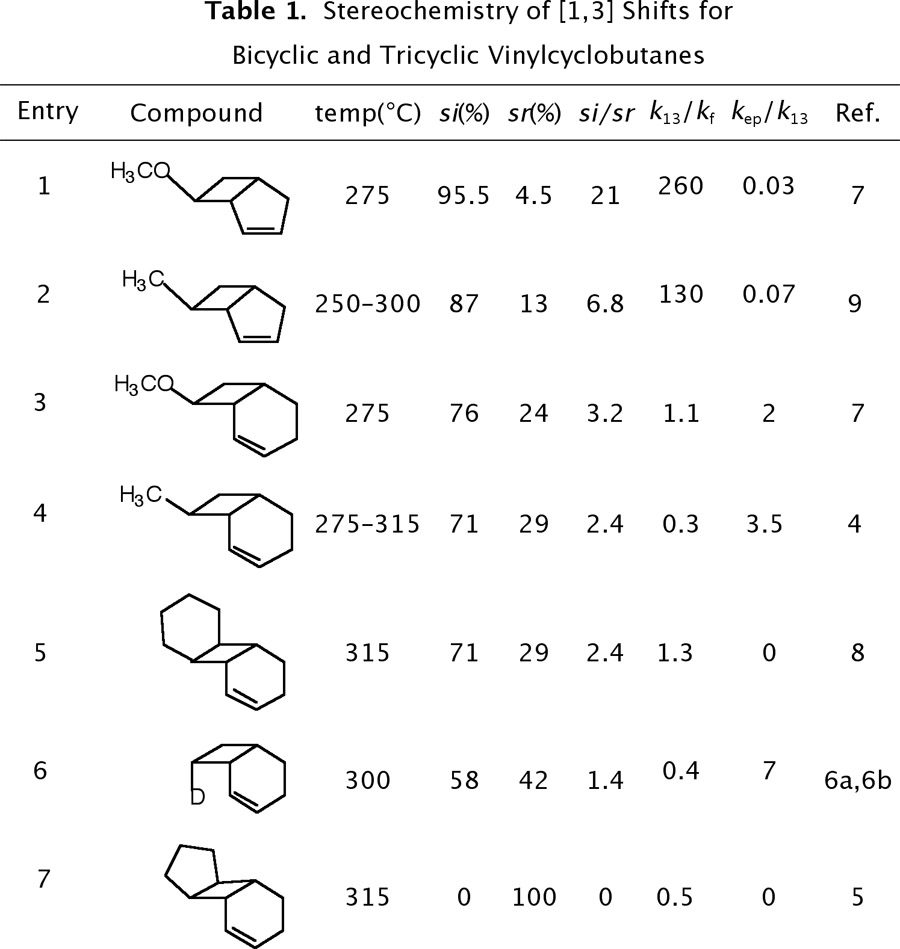
Bicyclo[4.2.0]oct-2-enes4-8 exhibit dramatically different thermal behavior compared to their corresponding bicyclo[3.2.0] homologs.7,9 In brief, one-centered stereomutation at the migrating carbon (kep) and fragmentation (kf) are generally more important than [1,3] carbon migration (k13), which is still stereoselective but only modestly so. We have recently completed three studies of bicyclo[4.2.0]oct-2-enes with the following trends (Table 1): 0.3 ≤ k13/kf ≤ 1.1, 2 ≤ kep/k13 ≤ 7, and 1.4 ≤ si/sr ≤ 3.2. The two tricyclic vinylcyclobutanes differ most markedly from their bicyclic analogs in that epimerization is nonexistent. We are currently investigating the thermal reactions of the 13-carbon tricyclic [4.2.0] analog (identified by the acronym 13TCT).
The relative contribution of the [1,3] rearrangement, one-centered stereomutation, and fragmentation processes in each bicyclo[4.2.0]oct-2-ene varies as a function of the extent of rotational torque accessible to the migrating carbon. The equivalence in the si/sr ratios for the fourth and fifth entries in Table 1 is probably not coincidental but rather an experimental measure of the inherent conformational flexibility imposed on the migrating carbon in [4.2.0] olefins where the migrating carbon carries an exo alkyl substituent. Houk has identified two vinylcyclobutane conformational interconversions as endo- and exo-ring openings which give si and sr products respectively.10 These observations further illustrates that dynamic factors, not orbital symmetry, control the thermal behavior of vinylcyclobutanes.
Impact Statement
Our research on bicyclic and tricyclic vinylcyclobutanes, the results of which are of interest to those engaged in computational dynamic modeling studies, and our collaboration with John Baldwin at Syracuse University have served as a stimulus to our scholarly productivity. Nine different F&M undergraduates, all of whom are currently in graduate school pursuing a Ph.D. in chemistry, have been coauthors on recent research papers acknowledging current PRF funding. All eight of these papers, seven of which appear in ACS journals, have been published in reputable venues.
References
(1) Woodward, R. B.; Hoffmann, R. The Conservation of Orbital Symmetry; Verlag Chemie: Weinheim, 1970.
(2) Leber, P. A.; Baldwin, J. E. Acc. Chem. Res. 2002, 35, 279-287.
(3) (a) Carpenter, B. K. J. Am. Chem. Soc. 1995, 117, 6336-6344. (b) Carpenter, B. K. Angew. Chem. Int. Ed. 1998, 37, 3340-3350.
(4) Bogle, X. S.; Leber, P. A.; McCullough, L. A.; Powers, D. C. J. Org. Chem. 2005, 70, 8913-8918.
(5) Baldwin, J. E.; Bogdan, A. R.; Leber, P. A.; Powers, D. C. Org. Lett. 2005, 7, 5195-5197.
(6) (a) Baldwin, J. E.; Leber, P. A.; Powers, D. C. J. Am. Chem. Soc. 2006, 128, 10020-10021. (b) Powers, D. C.; Leber, P. A.; Gallagher, S. S.; Higgs, A. T.; McCullough, L. A.; Baldwin, J. E. J. Org. Chem., 2007, 72, 187-194.
(7) Leber, P. A.; Lasota, C. C.; Strotman, N. A.; Yen, G. S. J. Org. Chem. 2007, 72, 912-919.
(8) Leber, P. A.; Bogdan, A. R.; Powers, D. C.; Baldwin, J. E. Tetrahedron 2007, 63, 6331-6338.
(9) Bender, J. D.; Leber, P. A., Lirio, R. R.; Smith, R. S. J. Org. Chem. 2000, 65, 5396-5402.
(10) Northrop, B. H.; Houk, K. N. J. Org. Chem. 2006, 71, 3-13.