www.acsprf.org
Reports: DNI150330-DNI1: Arylalkoxylation of Alkynes and Olefins via Transition Metal-Catalyzed C-O Activation
With support from the PRF, we have made considerable progress in utilizing transition metal-catalyzed activation of C–O bonds to enable rapid generation of molecular and stereochemical complexity from simple, readily accessible starting materials. As described in our proposal, we initially focused on developing new reactions via activation of aryl C–O bonds. Although these reactions have proven challenging, graduate students Andrew Ehle and Qi Zhou have begun to make progress in this area and will report our initial successes in this area soon. Our group has also begun to consider substrates with weaker C–O bonds for transition metal-catalyzed reactions and has found this to be a particularly interesting area for the discovery of new reaction technology.
In one area, we have discovered a novel transition metal-catalyzed reaction of N-benzoylaminals. Such aminals are attractive starting materials that can be easily accessed from acid chlorides, aldehydes and primary amines. However, few methods report the use of transition metal catalysts with aminals despite the potential for novel reaction pathways. Danielle McAtee, who joined this project as a first-year graduate student, has discovered that the use of a nickel(0) catalyst in conjuction with Lewis acid enables efficient cyclization of N-benzoyl aminals. This cyclization results in formation of 3-substituted isoindolinones, a molecular structure found in a number of biologically active molecules and natural products, including the anxiolytic pagoclone and natural pestalachloride A. With graduate student Srimoyee Dasgupta, Danielle has optimized the reaction conditions and determined that this reaction can be used to form a variety of these heterocyclic products.
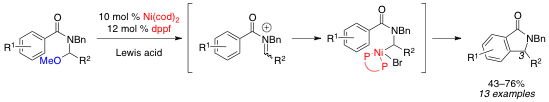
Intrigued by the role of nickel in this cyclization, we have pursued mechanistic studies of this reaction. The results of these studies suggest that the nickel catalyst is involved in the C–C bond formation and not in the C–O bond cleavage. Lewis acid alone is sufficient to reversibly form the iminium ion intermediate. Based on our studies and precedent from the Arndtsen group in related palladium chemistry, we propose that the electron-rich nickel catalyst reacts with the iminium ion to form an alpha-amidoalkylnickel(II) intermediate, which then likely reacts via either electrophilic aromatic substitution (EAS) or a migratory insertion pathway. Based on the observed substituent effects, we currently favor the EAS pathway. Our discovery, optimization, substrate scope and mechanistic experiments have been published recently and the publication added to the ACS-PRF web reporting system.
Our discovery of the nickel(0)-catalyzed cyclization of aminals has captured our imagination and inspired us to consider the possibility of related transformations, in which a transition metal catalyst controls the reactivity (or selectivity) of a cationic intermediate. To this end, we have begun a program in enantioselective, transition metal-catalyzed additions to prochiral oxocarbenium ions, formed in situ from racemic acetals. In particular, we are interested in enantioselective additions to cyclic oxocarbenium ions, because the requisite acetals are readily available by reduction of lactones and alpha-substituted oxygen heterocycles comprise a number of biologically active natural and synthetic products. Within this funding period, postdoctoral fellow Dr. Prantik Maity has developed a copper(I)-catalyzed alkynylation of isochroman acetals. By using a chiral bis(oxazoline) ligand, high enantioselectivies of the 2-alkynylisochromans are observed. To our knowledge, this is one of only 4 reported methods for enantioselective additions to cyclic oxocarbenium ions. Our report of this alkynylation is currently in press and will be added to the ACS-PRF web reporting system soon.
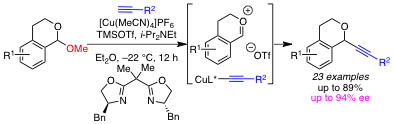
Our current studies are focused on the further development of this alkynylation and related chemistry. One of our highest priorities is to generalize this alkynylation to other acetal substrates, and we have preliminary results suggesting that enantioselective alkynylations of chromene acetals will also be possible. We are pursuing mechanistic analysis, as well as optimization studies, to reach this goal.
We are very excited to pursue these research directions and are grateful for the funding provided by the ACS PRF, which allowed us to develop this chemistry.