www.acsprf.org
Reports: DNI448755-DNI4: Photochemistry of Environmentally Relevant Nitro-Polycyclic Aromatic Hydrocarbons
The photochemistry of nitro polycyclic aromatic hydrocarbons (nitro-PAHs) continues to receive increased attention. This is in part due to the identification of nitro-PAHs as persistent organic pollutants in the environment as well as to their distinctive nonradiative excited-state dynamics. Early work has shown that nitro-PAHs containing two to four aromatic rings have vanishingly small fluorescence quantum yields, as well as, triplet yields approaching unity in solution at room temperature. The efficient population of the triplet state increases the probability of product formation; some of which have shown higher toxicity and carcinogenesis than the parent compounds.
During the last two year, our group has focused on studying the electronic structure, steady-state photochemistry, and excited-state dynamics of 1-nitronaphtalene (1NN), 2-nitronaphthalene (2NN), 2-methyl-1-nitronaphthalene (2M1NN), 1-nitropyrene (1NP) and 9-nitroanthracene (9NA) in solution. Quantum-chemical calculations predict that the torsion angle in these nitro-PAHs is modulated by electrostatic forces acting between the lone pairs of the oxygen atoms on the nitro-group and the neighboring atoms on the aromatic moiety. In the case of 1NN and 1NP, a peri hydrogen atom forces the torsion angle away from zero degree (i.e., away from the planar conformation). A methyl group in the ortho position of 1NN, in addition to a peri hydrogen atom, forces the torsion angle to even greater values in 2M1NN. A similar result is obtained for 9NA compared to 2M1NN; the former molecule having two peri hydrogen atoms. 2NN does not have peri hydrogen atoms that can interact with the nitro-group, which explain its planar conformation in the ground state (see Figure 1).
The calculations further predict that there is a distribution of torsion angles in the ground state at room temperature for each nitro-PAH. Upon excitation, the distribution of torsion angles access different regions of configuration space in the S1 state potential energy surface in each molecule, which is expected to modulate the time-resolved and steady-state photochemistry of these nitro-PAHs. For instance, Figure 1 shows potential energy scans for 2NN, 1NN, and 2M1NN in acetonitrile. The distribution of torsion angles accessible in the ground state at room temperature is highlighted in gray. These calculations predict a branching in the relaxation of the S1 state of these nitronaphthalene derivatives to populate two main relaxation channels. The first, main decay channel connects the S1 state with a receiver, high-energy triplet state (Tn) that has significant np* character. The second channel involves conformational relaxation of the vertical S1 state to populate the relaxed S1 state. The relaxed S1 state has significant charge transfer and negligible oscillator strength in 1NN and 2M1NN, thus forming an intramolecular charge transfer state, while the relaxed S1 state in 2NN only has small charge transfer character and significant oscillator strength (Fig. 1). This is because the nitro-group becomes perpendicular to the naphthalene moiety in the relaxed S1 state of 1NN and 2M1NN, while it is parallel to the naphthalene moiety in the relaxed S1 state of 2NN.
The computational results, together with the steady-state photodegradation measurements and the femtosecond transient absorption experiments, have also revealed that the S1 state branching is primarily controlled by the energy gap between the S1 and Tn states, as well as, by the ground-state distribution of nitro-aromatic torsion angles that is thermodynamically accessible at room temperature in each nitro-PAH. In general, the closer the distribution is to zero degrees the higher the triplet quantum yield, while the lower the photodegradation yield. For instance, the triplet yield of 2NN is 0.83 while the triplet yield of 1NN is 0.63 in solution. Similarly, the photodegradation yield of 2M1NN is higher than that of 1NN, while 2NN is practically photo-inert. Taken together, the experimental and computational results obtained by our group during the last two years suggest an important link between conformational distribution before light absorption and the excited-state dynamics and photochemistry in nitro-PAH compounds having two to four aromatic rings.
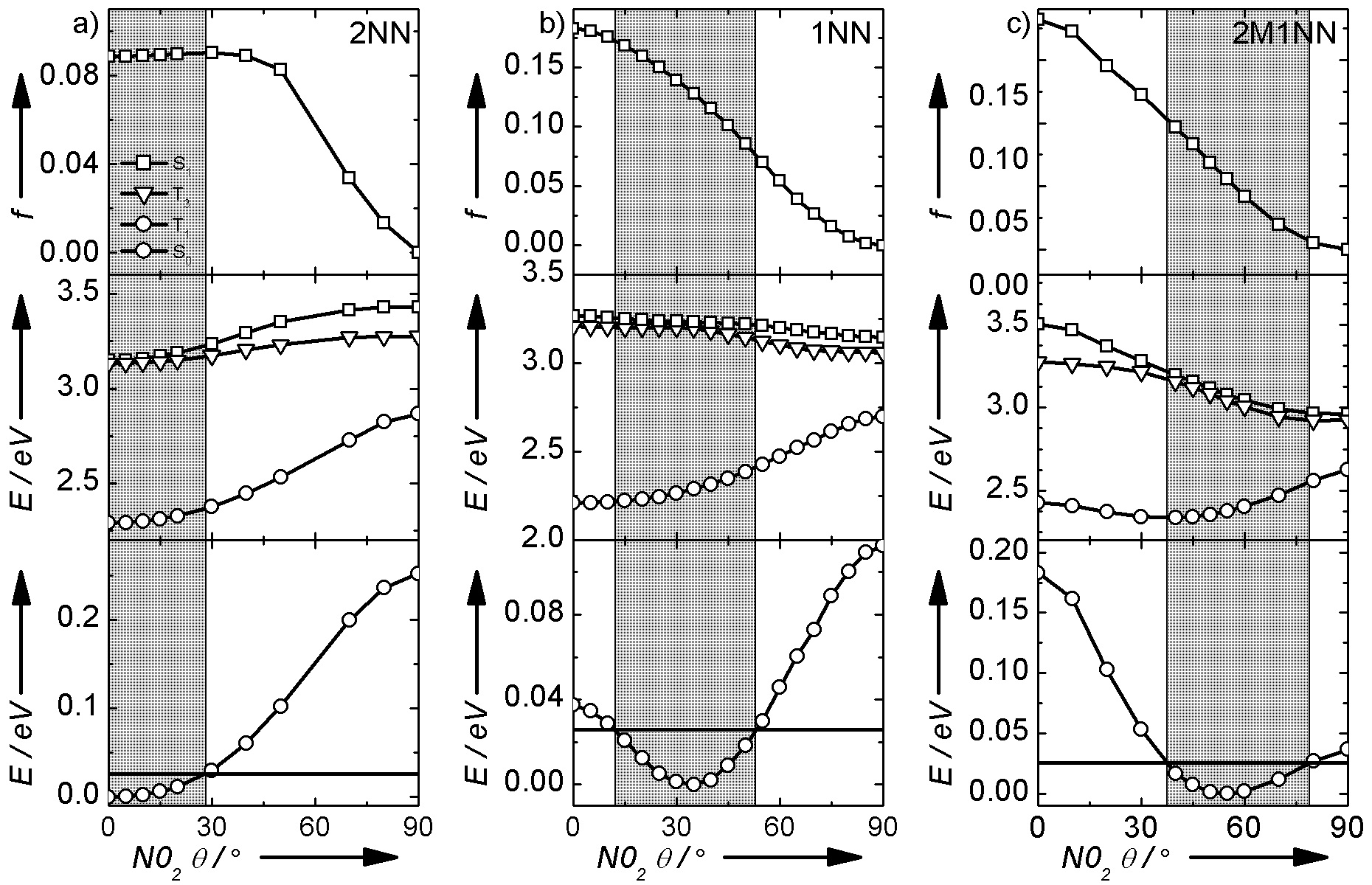
Figure 1. Potential energy scans for 2NN, 1NN, and 2M1NN as a function of the nitro-aromatic torsion angles in acetonitrile. The distribution of torsion angles accessible at room temperature is highlighted in gray. Vertical excitation energies were calculated at the TD-PBE0/IEPCM/6-311++G(d,p) level of theory following ground-state optimizations at B3LYP/IEFPCM/6-311++G(d,p) level of theory. The nitro-aromatic torsion angle was constrained to the specified values during the ground-state optimizations while all other parameters were unrestricted.