www.acsprf.org
Reports: ND348822-ND3: pi-Bonded Cationic Ligands as Catalysts and Precursors
Two years ago we were fortunate enough to be chosen for an ACS-PRF Type ND grant to investigate an entirely new concept – can pi-bonding ligands, which are normally either neutral or anionic in charge – be prepared in a cationic state? In the field of organometallic chemistry, there is perhaps no more important interaction than that which exists between a metal atom and the ligands that surround it, and much of the research in this field is driven towards understanding and controlling this interplay. As mentioned above, ligands commonly used in organometallic chemistry are either neutral or negative in charge. This is quite understandable, as ligands are Lewis bases that donate electron density towards the acidic metal. The nature of the charges on the ligands will determine what oxidation states are useful and can be utilized by the metal ion. However, it may be possible for ligands to be designed that have sufficient electron density to function as pi-donor Lewis bases, but also be cationic in charge. An illustrative scheme to prepare a P-C version of this class of compounds is shown in Fig. 1. This unique concept forms the basis of our work in this ND proposal, as we are aware only of sigma-bonding cationic ligands but not pi-bonding cationic coordinating ligands. The ligands that we have targeted are based on [(R2P)3C]+ or [(R2N)3C]+ tripodal frameworks, and most of our efforts to date have been directed towards the synthesis and characterization of the P-C based carbocation ligands. Previous work has shown that the neutral or anionic versions of these P- or N-based ligands exist and can coordinate to metals. We hypothesize that these cationic ligands should be stabilized by p(pi)-p(pi) bonding from lone pairs on the adjacent N or P heteroatoms. As such, we anticipate that these cationic ligands should bind to metal precursors to form new complexes. As we stated in our previous report, we have a unique opportunity to investigate a new family of ligands and to study them theoretically and experimentally, and most importantly, we have a possible opportunity to fundamentally alter the manner in which synthetic chemists think about ligand-metal interactions.
Fig.1 Scheme to Prepare P3C+ Ligand via Hydride Abstraction.
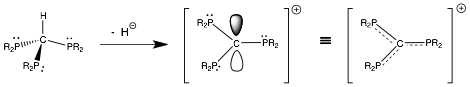
We have examined the concept computationally using DFT methods, and have shown that the cationic ligands should form attractive, net positive bonding complexes with metals. As well, when these tripodal cationic ligands were theoretically "bound" to metal fragments, three-fold symmetric pi-bound complexes were the lowest energy states. The N-C-N angles of the cationic ligand are ~120 degrees, thus indicating a flattening of the ligand framework that approaches planarity. Another indicator of pi-bonding interactions as shown by DFT is the shortening of the C-N bond.
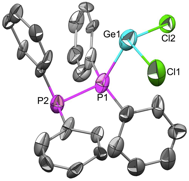
Synthetically, the removal of a hydride from the various starting hydrides has been challenging, to say the least. Much of our efforts to date have been directed towards the synthesis of the cationic ligands, in particular the [(R2P)3C]+ (R = Me, Ph) versions, beginning with the (R2P)3CH precursor. As documented in last year's report, we have had difficulty in removing the hydride. Use of acidic reagents such as HBF4, [Ph3C][BArf] (BArf = perfluorinated tetraphenyl borates), BPh3, AlMe3, B(C6F5)3, SbF5, TaF5, and the like did not show the ability to remove hydride from the starting (R2P)3CH species. The lack of hydride reactivity has been surprising to us, and has often resulted in the degradation of (R2P)3CH into the bis-species, (R2P)2CH2. This lack of desired reactivity has led us to attempt to prepare (R2P)3CX (X = halogen) derivatives, as halides are more effective leaving groups. Pure products have not yet been isolated, although new synthetic strategies have been developed for use during the final year of the project. We have prepared a number of neutral Group 6 metal complexes containing (R2P)3CH, and have attempted to remove the hydride from a "pre-coordinated" complex using strong acids. While this is not our desired method of preparing the cationic complexes, we are interested in the overall fundamental stability of these cationic complexes. While NMR evidence has indicated the removal of the hydride and the conversion into a cationic complex, we are cautious and apprehensive until we can obtain definitive single-crystal X-ray data for an authentic complex. A paper describing the neutral complexes is in preparation and will be submitted before the end of the calendar year. Work this final year will focus on both thermal and photochemical reactions of known [(R2N)3C]+ salts with neutral metal complexes containing labile ligands. As well, we are moving to heavier congeners of carbon to exploit the weaker E-H and E-X bonds found further down Group 14. In initial reactions to prepare (Ph2P)3ECl (E = Ge, Sn), we have observed unexpected reactivity of [Ph2P]Li with ECl4 to prepare not the desired compound, but rather a reductive coupling reaction to form Ph2P-PPh2 and ECl2. Interestingly, we have obtained structures of the mixtures of co-products. In the SnCl2 case, no interaction is seen between SnCl2 and Ph2P-PPh2 while in the GeCl2 case there is complexation in the solid-state with a P-Ge bond distance of 2.863 angstroms (shown in Fig. 2). The mechanism of this reaction is not yet known, but it appears excess lithium reagent is required.
Fig.2 Structure of GeCl2・(Ph2P-PPh2).
This grant from the ACS-PRF has been critical in getting our group involved in an entirely new area. As well, it has been instrumental for the training of two minority students – Dr. Wilson William and Dr. Agnes Mrutu – towards their Ph.D. degree or postdoctoral studies. While progress over the last year has been unfortunately hampered by a) graduation of the student working on this project, and b) scientific difficulties in generating the desired cationic complexes, a new postdoctoral fellow – Dr. William Pearl – from the laboratory of the highly-regarded professor Richard Adams at South Carolina has joined our group to attack this problem. We expect significant progress will be made on the identified areas during the final year.