www.acsprf.org
Reports: UNI149471-UNI1: Aziridination and Retro-Aldol Fragmentation of Dioxenones: Application in the Synthesis of alpha-Amino Acids and 2-Benzazepine Derivatives
Project Overview
Currently, our research group is pursuing the development of efficient methods to incorporate nitrogen into structurally complex and somewhat unstable 1,3-dioxenones via direct aziridination. Our plan is to use nucleophilic ring opening to manipulate the aziridine products into a variety of non-proteinogenic amino acids. More specifically, this project includes the simple construction of a variably substituted b-ketoester, such as 1, that is covalently trapped in its enolic form as a 1,3-dioxen-4-one (2). The development of an aziridination protocol for the dioxenones, then investigation of the retro-aldol fragmentation of the resultant bicyclic structure will furnish an array of substituted amino acids.

Numerous synthetic examples of aziridination are reported in the chemical literature, however there are few reports of aziridination of enol ethers, a,b-unsaturated esters and even fewer examples involving both the enol and ester functionalities in the same molecule, such as the 1,3-dioxen-4-one with which we are working. Known methods for the direct aziridination of alkenes are limited by the substitution of the alkene itself and the reactivity of the nitrogen equivalent. For example, many of these nitrogen equivalents are utilized as a high-energy nitrene intermediates that are rapidly added to an available alkene. The generation of nitrenes can be achieved by photolysis or thermolysis of an azide, or the formation of a metal-nitrene from iminoiodinanes, among other methods. Most of these methods are confined to sterically available alkenes and yields are calculated on the equivalency of the nitrene when using excess amount of the alkene substrate.
In this project, we have been directing most of our energy on the aziridination of dixoenone and pyranone substrates (such as 2). This current progress of this work, for the funding period of September 2010 to August 2011, is summarized below.
Summary of Experimental Work:
During the second year of this research, we have continued our investigation the direct aziridination of 2,2,6-trimethyl-1,3-dioxen-4-one (4). We had elected to begin our studies with dioxenone (4) due to its commercial availability. Our trials began with metal nitrene insertion to afford compound 5. Our initial result was promising, however after an exhaustive examination of this reaction, we found no possibility of reproducibility or acceptable yields using metal nitrenes formed from iminoiodinanes. Three different aryl iminoiodinanes were prepared, and a sampling of Rh2(OAc)4, Cu(I) and Cu(II) catalysts were tested. We also screened a number of conditions by changing both solvents and temperature, yet we never observed the complete consumption of the dioxenone 4 and we were also unable to isolate any appreciable amount of aziridine 5 that we once observed in the 1H NMR of the reaction mixture.
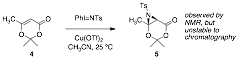
In an effort to develop a more atom economical approach to the aziridination, we focused on employing ethylazidoformate as a nitrene precursor to affect our desired aziridination. With precedent existing for nitrene formation under photolytic or thermolytic conditions (no metal catalyst required), we determined that aziridine 6 could be observed in about 30% conversion.
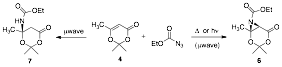
Using microwave irradiation, the reaction duration was minimized to one hour (also avoiding some thermal decomposition) and we were able to cleanly isolate and identify both products 6 and 7 of the reaction of ethylazidoformate with dioxenone 4 in dichloromethane (DCM), albeit in low yields. By changing the reaction solvent to a higher boiling solvent, dichloroethane (DCE), we retained the same results, but were unable to increase the yield of 6 in this transformation. We would still like to deduce the mechanistic pathway that accounts for the formation of carbamate ester 7 in these reactions. If 7 is a secondary product from the ring opening of the aziridine, we may be able to optimize the formation of 6 .
In addition to examining the reactivity of methyl-substituted dioxenone 4, our research group planned and synthesized two other dioxenone substrates with variable alkene substitution for use in the aziridination studies. The first of the derivatives, 8, was designed to probe the reactivity of a disubstituted alkene within the dioxenone ring. We had hoped that the pi system would be more sterically available to nitrene addition, however with less substitution, the dioxenone was also more prone to cycloreversion.
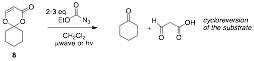
The second substrate, 9, replaced the methyl group with and phenyl ring. This change allowed us to have the stability of a trisubstituted alkene, but with different steric and electronic demands on the pi system. Product 10 was formed in moderate yield and the reaction showed little to no evidence of the aziridine opening or carbamate ester product like 7. We are now in the process of tuning the phenyl ring with both electron poor and electron rich substitutents to optimize this reaction.
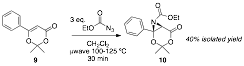
Concurrent with the work above, we prepared a seemingly more stable, pyranone substrate (11) to test our ethylazidoformate aziridination protocol. In just one aziridination attempt, the analogue showed conversion to the desired aziridine product. Even though 12 will not allow us the facile retro-aldol fragmentation in our original plan, we would like to optimize this result to include with our aziridination studies.
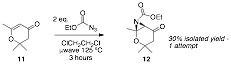
Conclusion and Future Direction
The details of this work were presented at the Local Mid-Hudson American Chemical Society Meeting on April 15th, 2011 at Bard College by undergraduates who have contributed to various aspects of this project. These Bard College undergraduates include – Madison Fletcher '12, Youseung Kim '12, Nicole Camasso '12, and Nathan Steinauer '13. Prabarna Ganguly '13 and Xiaohan Sun '13 also contributed to the synthesis of dioxenone derivatives.
This project has led to the development of a research program concerning the dirhodium catalyzed intramolecular aziridination to prepare both dihydroisoquinolones and dehydroisoindolone scaffolds.
At this time we are in the process of reproducing our current data, generating parallel examples, and testing variably substituted dioxenones toward publication (with undergraduate coauthors) within the year.