www.acsprf.org
Reports: DNI350287-DNI3: Physical Properties and Reactivities of Peroxomanganese(III) Complexes
The subject of this award is to contribute to an in-depth understanding of the initial steps in hydrogen peroxide activation for manganese catalysts by defining the physical and reactivity properties of peroxomanganese(III) intermediates, which are proposed to be among the first species formed when manganese complexes react with hydrogen peroxide. Such catalytic systems are of interest because manganese centers and hydrogen peroxide are able to mediate a variety of important oxidative transformations, including those involved in the functionalization of petroleum feedstocks and fabric and paper bleaching. In year one of this project we have generated a series of peroxomanganese(III) complexes and have used a variety of spectroscopic and computational tools to relate systematic changes in the properties of the supporting ligand to changes in reactivity and electronic structure.
A series of peroxomanganese(III) complexes supported by tetradentate, aminopyridyl and aminoquinolinyl ligands (Scheme 1) were generated by treatment of manganese(II) precursor complexes with hydrogen peroxide and base (Figure 1). As shown in Figure 1, electronic absorption spectra of all peroxomanganese(III) compounds display features in the visible region at ~17 000 and 22 000 cm-1. These complexes are metastable, with half-lives varying from ~5 seconds to 15 minutes at 0 °C. The relative reactivities of the more stable complexes were compared using the deformylation of cyclohexanecarboxaldehyde as a benchmark reaction. Deformylation rates were strongly dependent on ligand sterics. For example, the [Mn(O2)(L7py2H)]+ complex, which lacks any pyridine ring substituents, showed the fastest second-order rate of 3.1 M-1min-1,whereas the [Mn(O2)(L7py26-Me)]+ and [Mn(O2)(L7py24-Me)]+ complexes, which respectively contain methyl substituents in the six and four positions of the pyridine rings (Scheme 1), were an order of magnitude slower (0.32 and 0.40 M-1min-1, respectively). Moreover, on the basis of comparison to other peroxomanganese(III) species, [Mn(O2)(L7py2H)]+ is the fastest peroxomanganese(III) deformylating agent lacking an anionic ligand. These studies were reported in Dalton Transactions in 2011. In year two of this project, these experiments will be extended to a wider range of organic substrates.
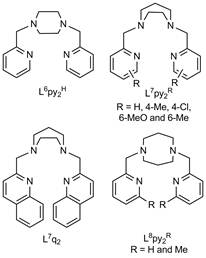
Scheme 1. Structures of aminopyridyl and aminoquinolinyl ligands used to support peroxomanganese(III) complexes.
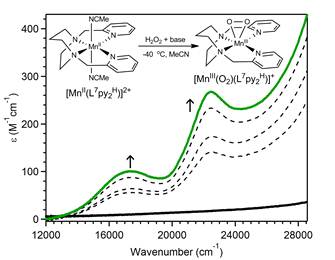
Figure 1. Electronic absorption spectra showing the formation of [MnIII(O2)(L7py2H)]+ from [MnII(L7py2H)(MeCN)2]2+ using H2O2 as oxidant. The inset shows a schematic representation of the reaction chemistry.
The electronic structures of these peroxomanganese(III) complexes were characterized by complementing our electronic absorption data with magnetic circular dichroism (MCD), and variable-temperature, variable-field (VTVH) MCD data. Analysis of the VTVH MCD data revealed similar ground-state zero-field splitting parameters, consistent with the formulation of all species as side-on peroxomanganese(III) adducts. However, the electronic transitions observed in the MCD spectra show important variations among this series. Most notably, steric bulk in the six position of the pyridine ring is correlated with a red-shift of the lowest-energy MnIII d-d transition. On the basis of our previous work (Geiger, et al., J. Am. Chem. Soc., 2010, 132, 2821.), the energy of this transition is a marker for the strength of the Mn-peroxo interaction, with a lower-energy transition being indicative of a weaker interaction. Surprisingly, essentially no variation in electronic transition energies was observed for peroxomanganese(III) complexes supported by the L7py24-Cl, L7py2H, and L7py24-Me ligands, which have systematically perturbed electronic properties (Figure 2). Instead, large spectral changes were observed between peroxomanganese(III) complexes supported by the L7py26-Me and L7py24-Me ligands (Figure 2). These data clearly demonstrate that it is the location, and not the identity, of the pyridine ring substituent that influences Mn-peroxo bonding. The steric properties of the supporting ligand can play a dominant role in modulating metal-peroxo bonding by influencing the size of the peroxo binding pocket. A paper covering these spectroscopic studies and complementary computational analysis is in preparation.
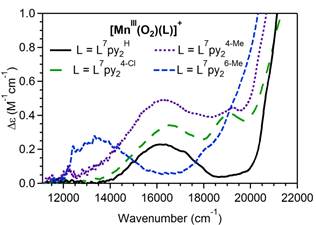
Figure 2. 4 K, 7 T MCD spectra of [MnIII(O2)(L7py2R)]+ complexes (R = H, 4-Me, 4-Cl, and 6-Me) showing the lower-energy region that contains the lowest-energy d-d transition.
Using this powerful spectroscopic and computational methodology, we investigated the geometric and electronic structures of peroxomanganese(III) complexes supported by the pentadentate mL52, imL52, and N4py ligands (Scheme 2, top) generated in our lab and in the lab of Dr. Elodie Anxolabéhère-Mallart (Université Paris Diderot, Paris, France). We note that [MnIII(O2)(imL52)]+ is the first peroxomanganese(III) complex containing biologically-relevant imidazole ligands. All three peroxomanganese(III) complexes are quite unstable at temperatures above -40 °C, and this has hampered the use of X-ray crystallography for gaining structural insights into these complexes. This is clearly a major drawback in understanding reactivity, which include activation of the peroxomanganese(III) unit with acid (Groni, et al., Inorg. Chem. 2008, 47, 3166.). Given the lack of crystallographic data, a number of structures have been proposed (Scheme 2). To address this issue we used spectroscopic data to serve as a basis for distinguishing several hypothetical models developed using DFT computations. For example, Figure 3 shows the experimental absorption spectrum of [MnIII(O2)(imL52)]+ as well as a computed spectrum using a model with a side-on peroxo ligand and with the mL52 ligand bound in an unusual tetradentate fashion. On the basis of computed energies and spectroscopic data, we concluded that the peroxomanganese(III) complexes supported by mL52, imL52, and N4py ligands consist of six-coordinate MnIII centers with side-on peroxo ligands and tetradentate supporting ligands. Complementary computations on analogous peroxoiron(III) complexes supported by the same ligands showed that a seven-coordinate geometry is favored for the Fe systems, which stands in direct contrast to that proposed for the Mn compounds. These studies show the coordination preferences of FeIII- and MnIII-peroxo species are distinct, which has implications for Fe and Mn dioxygenase enzymes that are often proposed to share a common mechanism. A paper concerning this work has recently been published (On-line ASAP) in Inorganic Chemistry.
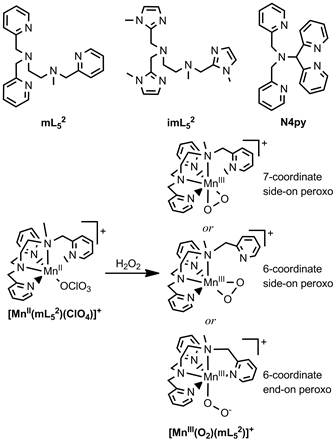
Scheme 2. Top: Pentadentate ligands used to support peroxomanganese(III) complexes. Bottom: Formation of [MnIII(O2)(mL52)]+ from [MnII(mL52)(ClO4)]+ including representations of three possible structures of the product.
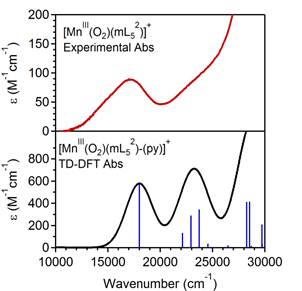
Figure 3. Top: Experimental electronic absorption spectrum of [MnIII(O2)(mL52)]+. Bottom: Computed absorption spectrum using time-dependent DFT computations and a model of [MnIII(O2)(mL52)]+ with a side-on peroxo ligand a dissociated pyridine group from the mL52 ligand.