www.acsprf.org
Reports: UNI349494-UNI3: Electronic Tuning of Magnetic Exchange in Phenoxy-bridged Dinuclear Transition Metal Complexes
Introduction
Schiff base macrocycles provide ideal scaffolds to study transition metal coordination. Schiff base macrocycles are synthesized through the transition metal templated condensation reaction of 2,6-diformylphenols and diamine linkers, Figure 1. The linking arm can be changed as necessary to accommodate the different coordination needs of the different transition metal ions.
The binding pocket created by the bridging phenols and the imine linkers is ideal for the controlled coordination of a variety of different transition metals. Their synthesis is straightforward and amenable to variety of structural modifications through substitution on the aryl rings or changing the identity of the linking amine bridge. Research in our lab has been aimed at elucidating how magnetic exchange of transition metal ions is affected by altering the electron density of the phenoxy bridge through substitution on the aryl ring.
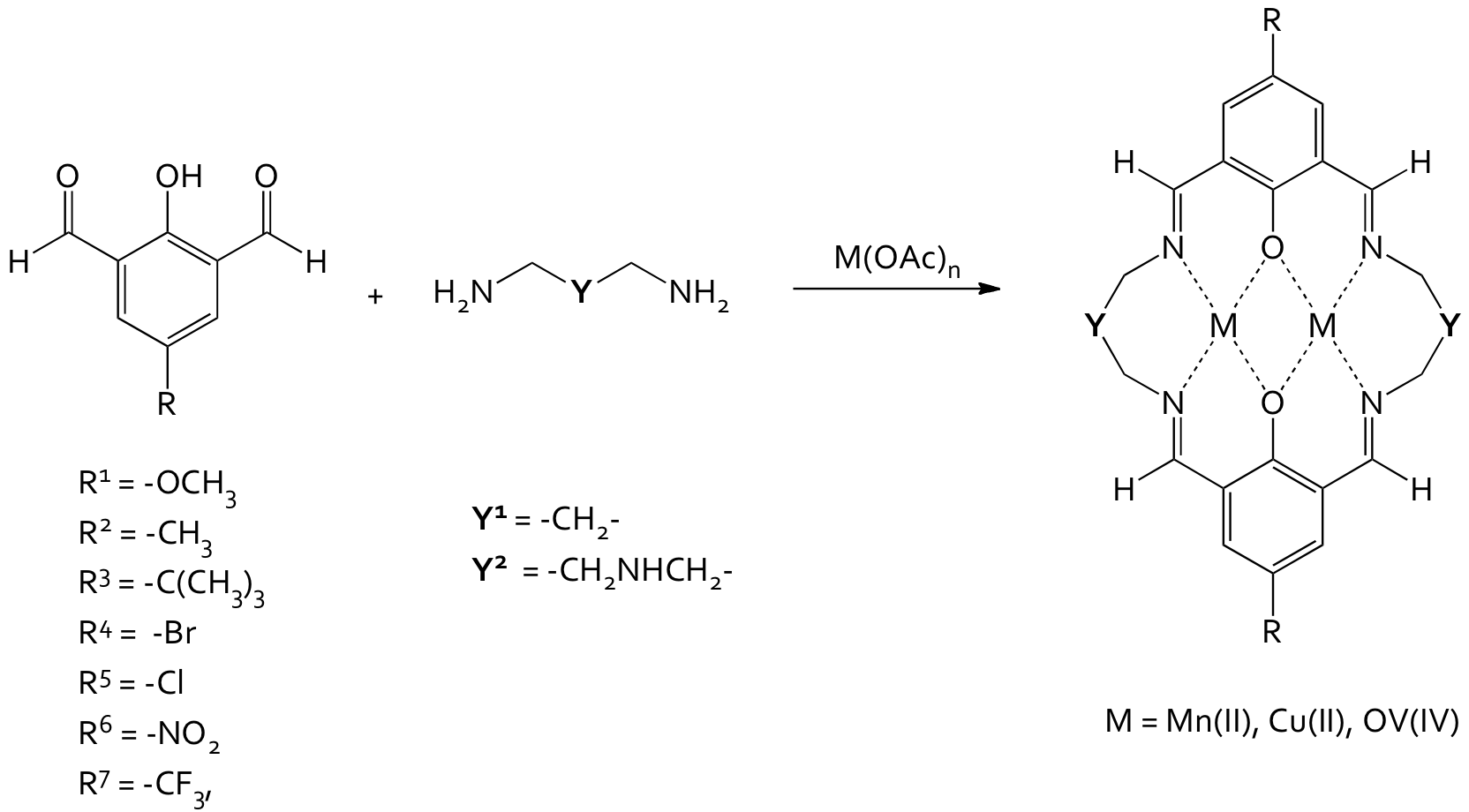
Figure 1: General synthetic scheme for Schiff base macrocycles.
A series of complexes ([M2YR], where M = transition metal ion, L = linking arm and R = substituent on aromatic ring) was synthesized and their magnetic exchange parameters measured in order to correlate the strength of the measured magnetic exchange (J) with the Hammett constants of the substituent. Substituents were chosen to span the range of electron-withdrawing and electron-donating abilities to give the broadest range of Hammett constants.
Results
Synthesis of Dialdehyde Precursors
In order to synthesize the macrocyclic complexes, we must first make the diformyl precursors using the Duff formylation, Figure 2. This reaction gives both the mono- and diformyl products (shown in Figure 2). In the previous granting period (2009-2010), we developed a microwave procedure that gave comparable yields to traditional reflux methods and favored the formation of the diformyl product.
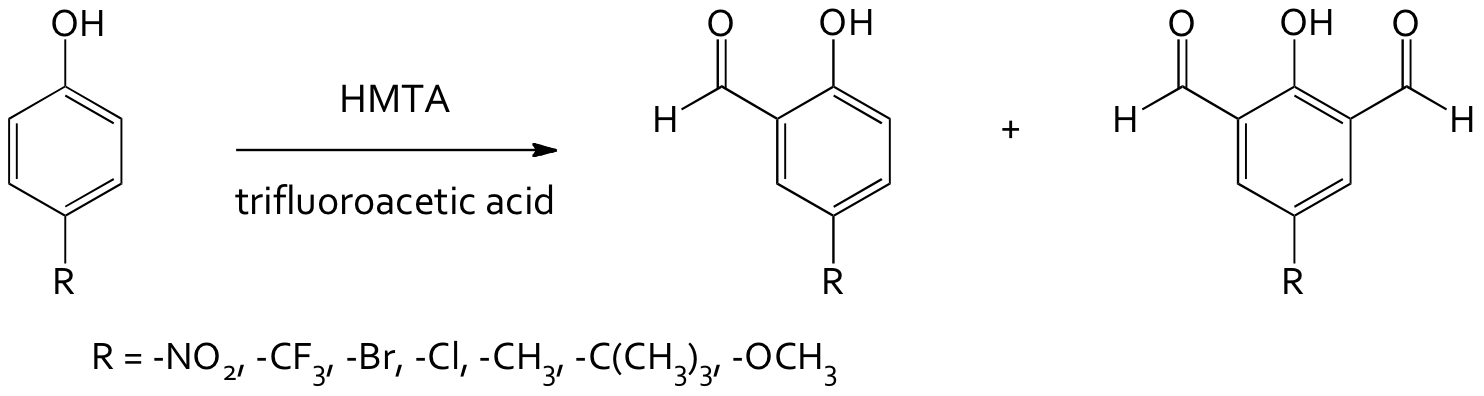
Figure 2: Duff formylation reaction of para-substituted phenols.
During the past year, several undergraduate students have continued to work on increasing overall yields and the amount of the diformyl product. We have been investigating the use of solid supports and stronger acid catalysts.1 In contrast to previously reported reactions, the use of silica gel and zeolite supports did not produce useable products. There was a large amount of degradation and polymer formation.
Dimanganese(II) Complexes
In the previous year (2009-2010), we synthesized and characterized the [Mn2Y1R] complexes with R2-R5. The magnetic susceptibility of the complexes was measured and the value of J was determined and plotted against the Hammett constants but no correlations were identified. However, this set of substituents does not represent the extremes of the electron-donating and withdrawing groups.
During this granting period, we have been working to synthesize both the nitro and trifluoromethyl substituted Mn(II)2 complexes. These substituents are both highly electron withdrawing and represent one extreme of the series. The yields of these complexes is very low and is being hindered by our difficulties in purifying the –NO2 and –CF3 substituted starting materials. We have successfully synthesized the methoxy complex, but it is not stable in solution and decomposes rapidly during purification.
We have also been hampered in our ability to grow single crystals of these complexes. The crystals that we have been able to grow are thin sheets that are not suitable for x-ray analysis. We are currently investigating different solvent systems.
Copper(II) Complexes
We have also begun to investigate Cu(II) dimers using the same general macrocyclic structure. The switch to Cu(II)2 will reduce the number of magnetic exchange pathways that are possible. Copper(II) is d9 with the unpaired electron in the dx2-y2 orbital. This orbital should interact with the sigma* orbitals of the bridging phenoxy ligands. For the synthesis of the copper dimers, we have used 1,3-diaminopropane (Y2) as the linking arm. We have synthesized five of the complexes and obtained x-ray structures of the bromo, methoxy and tert-butyl complexes.
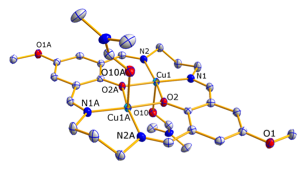
Figure 3: X-ray structure of [Cu2Y1R1](ClO4)2. Hydrogen atoms are omitted for clarity. Coordinated solvent molecules of DMF are shown.
Shown in Figure 3 is the crystal structure of the [Cu2Y1R1](ClO4)2 complex. The crystals were grown by slow diffusion of diethyl ether into a DMF solution of the dimer. [Cu2Y1R4](ClO4)2 was obtained in the same manner, while [Cu2Y1R3](ClO4)2 was grown from slow diffusion of diethyl ether into an acetonitrile solution.
We are working with Michigan State University at the current time to obtain magnetic susceptibility data on the copper complexes. We have time scheduled during October, 2011.
Current Work
We are currently working towards the synthesis of oxovanadium dimers. The vanadium is d1 with the unpaired electron in the lower energy d orbitals (dxz or dyz). We have had more difficulties with these syntheses, despite using published synthetic schemes.2 In order to overcome these difficulties, students been working on the synthesis and characterization of substituted salicylaldehye carbazone oxovanadium complexes to better understand the chemistry of oxovanadium complexes.3
Student Support
Funds from the have been used to support two undergraduates during Summer 2011. Three additional students (including two underrepresented minorities) have worked on aspects of this project during the semester as directed studies. These students have given poster presentations at national ACS meetings, and oral presentations at a campus wide research symposium.
References
1Yaseen, M.; Ali, M.; Ullah, M.N.; Munawar, M.A.; Khokhar, I. 2009. Journal of Heterocyclic Chemistry, 2009, 251-55.
2 a) Das, R.; Nanda, K.K.; Mukherjee, A.K.; Mukherjee, M.; Helliwell, M.; Nag, K. J. Chem. Soc. Dalton Trans. 1993, 22, 2241-2246. b) Mandal, S.K.; Nag, K. J. Org. Chem. 1986, 51, 3900-3902.
3Noblia, P; Vieites, M; Parajon-Costa, BS; Baran, EJ; Cerecetto, H; Draper, P; Gonzalez, M; Piro, OE; Castellano, EE; Azqueta, A; de Cerain, AL; Monge-Vega, A; Gambino, D. Journal of Inorganic Biochemistry. 2005, 99, 443-451.