www.acsprf.org
Reports: ND648890-ND6: Adsorption and Chromatographic Separation of Chain Molecules on Nanoporous Substrates
Abstract. Adsorption of chain molecules on nanostructured surfaces and within nanoscale pores is the key mechanism of chromatographic separation and characterization of polymers, which is one of the most widespread analytical techniques in the petroleum and petrochemical research and development. Chromatographic separation depends on the different types of physico-chemical interactions between the macromolecule and the surface of porous particles packed into a chromatographic column. The most efficient separation is achieved at the critical point of adsorption (CPA) that corresponds to a delicate balance between repulsive steric and attractive adsorption interactions coupled with confinement effects, which allows for characterization of macromolecules based on their chemical composition, microstructure, and topology, independent of molecular weight. A better understanding of the pore structure effects, such as pore size and shape, is needed for a rational design of chromatographic columns, choice of porous supports, and optimization of experimental protocols. The primary aim of the proposed program is to gain a fundamental understanding of the physico-chemical mechanisms of polymer adsorption in nanopores and the conditions of CPA based on the development of new methods for multiscale molecular simulations of real chromatographic systems.
The results of this proposal served as a foundation for the full-scale NSF-GOALI proposal that was awarded to the PI to continue studies of polymer chromatography with molecular simulation methods.
Results for the reporting period.
The main focus of the research was on the application of a new simulation
method, gauge cell mesoscopic ensemble Monte Carlo (MCMC), for modeling
equilibrium adsorption and dynamics of chain molecules in nanopores. This
method was found to be significantly more efficient than previous approaches.
We illustrated the practicality of MCMC by calculating the partition coefficient
between a polymer chain and a spherical adsorbing pore, and by using the
Focker-Plank equation to predict translocation dynamics from the free energy
landscapes obtained by our new method.
MC simulation (Christopher Rasmussen, PhD student, and Dr. Aleksey Vishnyakov, Research Assistant Professor). We have implemented the incremental gauge cell (IGC) mesoscopic Monte Carlo (MCMC) method for non-branched chains of Lennard-Jones (LJ) atoms. We were able to obtain a quantitative agreement with the results obtained by modified-Widom (MW) method of Kumar et al. (1992) for free (non-adsorbed) chains. The gauge cell allows us to “measure” the chemical potential in the considered system, as well as moderate any large density fluctuations, thereby stabilizing metastable and labile states. The free energy of various systems (unabsorbed, weakly adsorbed, etc.) can be compared to determine the most favorable state. We assessed the efficiency of our method by finding the number of MC steps required to arrive at a chemical potential with identical precision between the IGC and MW methods (Figure 1). The IGC method is found to be approximately 1 order of magnitude more efficient. Chains were simulated both free and in confinement (adsorbing and hard-walls). The chemical potentials of these systems are shown in Figure 2; the free energy is the sum of these values. Estimation of the chromatography partition coefficient K can be done by comparing the free energy of adsorbed and unconfined chains, K=exp(-ΔF/kT). Figure 3 shows the values of K for n-alkane chains with spherical silica pores. The IGC method was used to model polymer translocation through a small opening. This was done by tethering one end of the chain to a fixed point, and calculating the chemical potential of chains on a wall (e.g. the bulk) and in an adsorbing pore (e.g. the translocated polymer). The dynamics of translocation can then be modeled by using the difference in free energy between these two states in the Focker-Plank (FP) equation. Figure 4 shows this free energy landscape of partially translocated polymer chains, and Figure 5 displays the translocation time from the FP equation.
Figure 1. Comparison of the
computational efficiency between the incremental gauge cell and MW methods for
a single chain confined in a hard wall sphere of diameter 10σ at T*=1. The vertical axis represents normalized length of simulation
required to reach an acceptable level of errors in chemical potential
calculations chosen as ±0.01kBT.
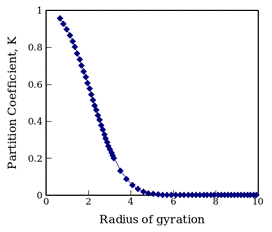
Figure 3. The partition coefficient between an LJ chain in a
good solvent (e.g., T*=8) and an adsorbing
spherical pore of R=5σ, plotted
against the radius of gyration of an unconfined chain. The parameters used
correspond approximately to n-alkane
adsorbing on silicia. Such an adsorbent would operate in the size-exclusion
regime, since K is always less than
one. All chains larger than RG~5σ will not adsorb and would elute
with the solvent band in a chromatographic separation.
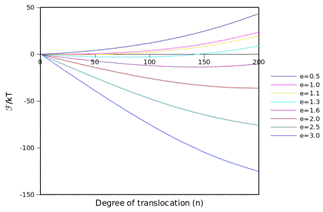
Figure 4. The free energy landscape of a translocating
chain, determined by the incremental gauge cell method. F is
the difference in free energy per degree of translocation, and e is the strength of the adsorption
parameter relative to the monomer-interaction parameter. For short chains, e must be larger than 1 (e.g., the
monomer-pore interaction must be larger than the monomer-monomer interaction).
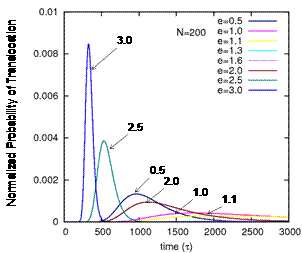
Figure 5. First passage times of a translocating chain of
200 monomers into a spherical pore, obtained by application of the Focker-Plank
equation to the free energy results in Figure 4 above. A strong adsorption
parameter is found to facilitate fast translocation.
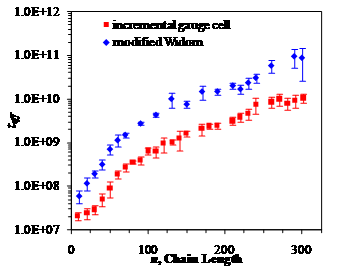
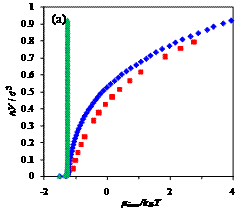
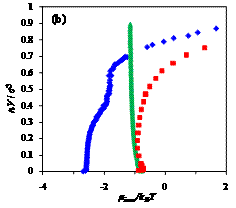
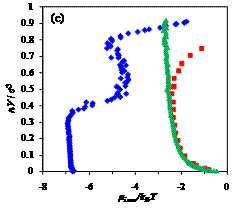
Figure 2. Dependence of the
incremental chemical potential on chain length n of free, confined, and adsorbed chains at different temperatures,
T*= (a) 8, (b) 2, (c) 1;
blue diamonds (♦) - chain in an
adsorbing pore of volume V/σ3=523,
red squares (■) - chain in the same pore
with only hard-wall repulsion, and green triangles (▲)
- free chain (no confinement). At high temperatures, entropic factors dominate
and chemical potential sharply increases with chain length. Conversely, at low
temperatures, enthalpic interactions are most important: the adsorbing chain
can be observed in layered configurations, and the free chain is condensed into
a globule.