www.acsprf.org
Reports: DNI950202-DNI9: Molecular Dynamics Simulation of Surfactant-Driven Wettability Alteration of Calcium Carbonate for Enhanced Oil Recovery
Narrative Progress Report
Background
Since ~ 50 % of the petroleum reserves are found in carbonate reservoirs and the average oil recovery from carbonate reservoirs through the water flooding method is often less than 30 %, the development of more efficient and productive technology for enhanced oil recovery (EOR) is very essential to fully utilize our energy resources as well as to reduce the energy consumption in producing a unit volume of petroleum from the reservoirs. In order to enhance the oil recovery from carbonate reservoirs, it is desirable to alter the wettability of the carbonate surface using surfactants from the oil-wet surface to the water-wet surface. However, the detailed mechanism of the wettability alteration has not been understood completely at molecular level, which should be a critical requisite in developing better EOR technology. Thus, we are investigating the molecular mechanisms of the surfactant-mediated wettability alteration on the carbonate surface.
Objectives
- To predict probable structures of the carboxylate-adsorbed calcium carbonate surface using first principle density functional theory (DFT) calculations (1st year)
- To simulate the surfactant-driven desorption of carboxylate from the calcium carbonate surface using molecular dynamics (MD) simulation (2nd year)
Computational Methods
All the DFT
calculations in this study were performed using the Generalized
Gradient Approximation (GGA) density functional theory (DFT)
in the DMol3 with the Perdew-Burke-Ernzerhof
(PBE) functional. We used
all-electron Kohn-Sham wave functions and DNP numerical basis set for the
spin-unrestricted DFT computation. The
Brillouin zone is sampled by 3•5•4
Monkhorst-Pack (MP) k-point mesh.
The self-consistent field (SCF) convergence is obtained at the given k-point
sampling at 1.0•10-5 Ha. The lattice parameters
(a = 8.1 Å and b = 5.0 Å)
of calcite
![]() surface
which has been well characterized with experiment were used for this
calculation.
surface
which has been well characterized with experiment were used for this
calculation.
Results and Discussions
|
Figure 1. Model structure of calcite |
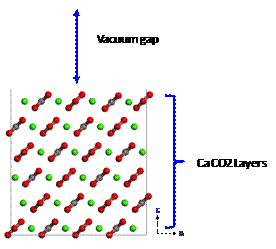
1.
Determination of calcite
![]() slab
structure
slab
structure
First of all, we determined the slab thickness and vacuum gap size of the model (Figure 1) by calculating surface energy (J/m2):
![]() (1)
(1)
where ![]() and A denote the surface
energy (J/m2) and the area, respectively. US and UB
are the energy of surface slab and the energy of bulk crystal, respectively.
We found that a slab consisting of six layers of calcite was sufficient (Table
1). The size of the vacuum gap showed no
effect on the surface energy as shown in Table 2. The gap of 100 Å was
adopted for this investigation.
and A denote the surface
energy (J/m2) and the area, respectively. US and UB
are the energy of surface slab and the energy of bulk crystal, respectively.
We found that a slab consisting of six layers of calcite was sufficient (Table
1). The size of the vacuum gap showed no
effect on the surface energy as shown in Table 2. The gap of 100 Å was
adopted for this investigation.
Table 1. Surface energy as a function of the calcite
|
Table 2. Surface energy as a function of the vacuum gap size
|
2.
Adsorption of calcium ion on the calcite ![]() slab
surface
slab
surface
|
Figure 2. Three different sites (A, B or C) for calcium ion adsorption on the calcite |
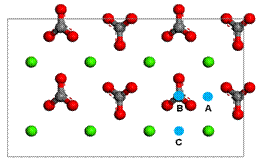
We used a periodic
slab structure with a = 16.2
Å
and b = 10.0
Å
to provide sufficient surface area to accommodate calcium ion and benzolate.
To investigate the adsorption configuration of calcium ion on the calcite
surface,
we set three different sites (A, B or C) for calcium ion as shown in Figure
2. After the geometry-optimization using DFT, we found that calcium ion
is moved to the one common site shown in Figure 3 regardless of their initial adsorption
sites (Figure 2). From this adsorption site, the adsorption
energy (![]() )
of calcium ion is calculated as -70.8 kcal/mol using the following definition:
)
of calcium ion is calculated as -70.8 kcal/mol using the following definition:
![]() (2)
(2)
where ![]() ,
, ![]() and
and ![]() denote the
energy of the calcite with the adsorbed calcium
ion,
the energy of calcium ion and the energy of calcite,
respectively.
denote the
energy of the calcite with the adsorbed calcium
ion,
the energy of calcium ion and the energy of calcite,
respectively.
3.
Adsorption of benzolates on the calcite
![]() surface
surface
|
|
Figure 3. The final adsorption position of calcium ion on the calcite |
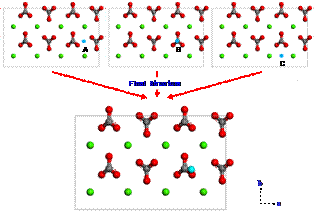
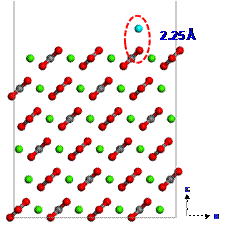
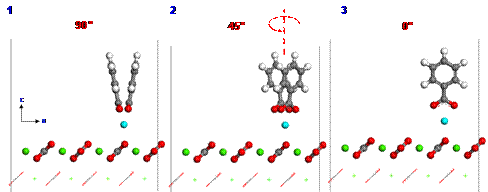
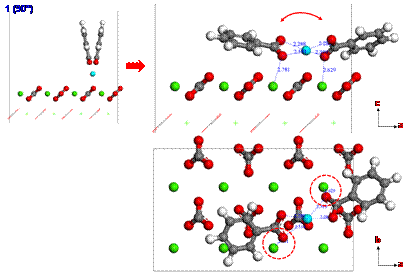
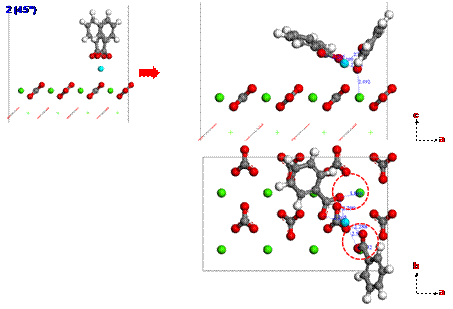
Since calcium ions are adsorbed from the brine on the calcite surface, carboxylates can be strongly interacted with calcium ions. To investigate the adsorption configuration of benzolates, we set three different configurations (1, 2 or 3) for benzolates adsorbed on the calcium ion as shown in Figure 4. We used two benzolates to neutralize a charge of the model system. The final structures from such different initial configurations are obtained using DFT geometry optimization as shown in Figures 5 to 7. Each oxygen of benzolate interacts with the adsorbed calcium ion strongly as well as the closest calcium atom on the first layer of calcite surface (see red circles in Figure 5 to 7). However, we found that the adsorption energy is not significantly dependent on the initial positions of benzolates as shown in Table 3.
Table 3. Adsorption energy per benzolate
|
4. Adsorption energy correction
In this study, we calculated the adsorption energy using PBE with the Grimme scheme. The Adsorption energy per benzolate was -84.0 kcal/mol, indicating that the adsorption of benzolate is a chemisorption.
|
Figure 4. Three different initial configuration (1, 2 or 3) for benzolates adsorbed on the calcium ion. . |
|
Figure 5. The adsorption configuration of benzolates from the initial structure 1 after geometry optimization using DFT. |
|
Figure 6. The adsorption configuration of benzolates from the initial structure 2 after geometry optimization using DFT. |
|
Figure 7. The adsorption configuration of benzolates from the initial structure 3 after geometry optimization using DFT. |
Perspectives
The
work enhances the knowledge of the adsorption of carboxylates and calcium ions on
the calcite ![]() surface
using DFT method. DFT calculations in this study will be used to optimize
the force field parameters, especially Morse potential energy functions for
large-scale molecular dynamics simulation for the next step of the project.
surface
using DFT method. DFT calculations in this study will be used to optimize
the force field parameters, especially Morse potential energy functions for
large-scale molecular dynamics simulation for the next step of the project.
Narrative Words: 992