www.acsprf.org
Reports: GB447864-GB4: Generation and Kinetic Studies of High-Valent Metal-Oxo Intermediates
Scientific and Technical Description of the Results
Photochemical generation of trans-dioxoruthenium(IV) porphyrin complexes
First, we report our full findings on the photochemical generation of trans-dioxoruthenium(VI) complexes (3) by irradiation of porphyrin-ruthenium(IV) dichlorate or dibromate complexes (2) that result in homolytic cleavage of the O-X bond in both axial ligands (Scheme 1). We have demonstrated that this photochemical method can be used to generate trans-dioxoruthenium(VI) species in six porphyrin systems under similar condition, in particular the very electron-demanding RuVI(TPFPP)O2.
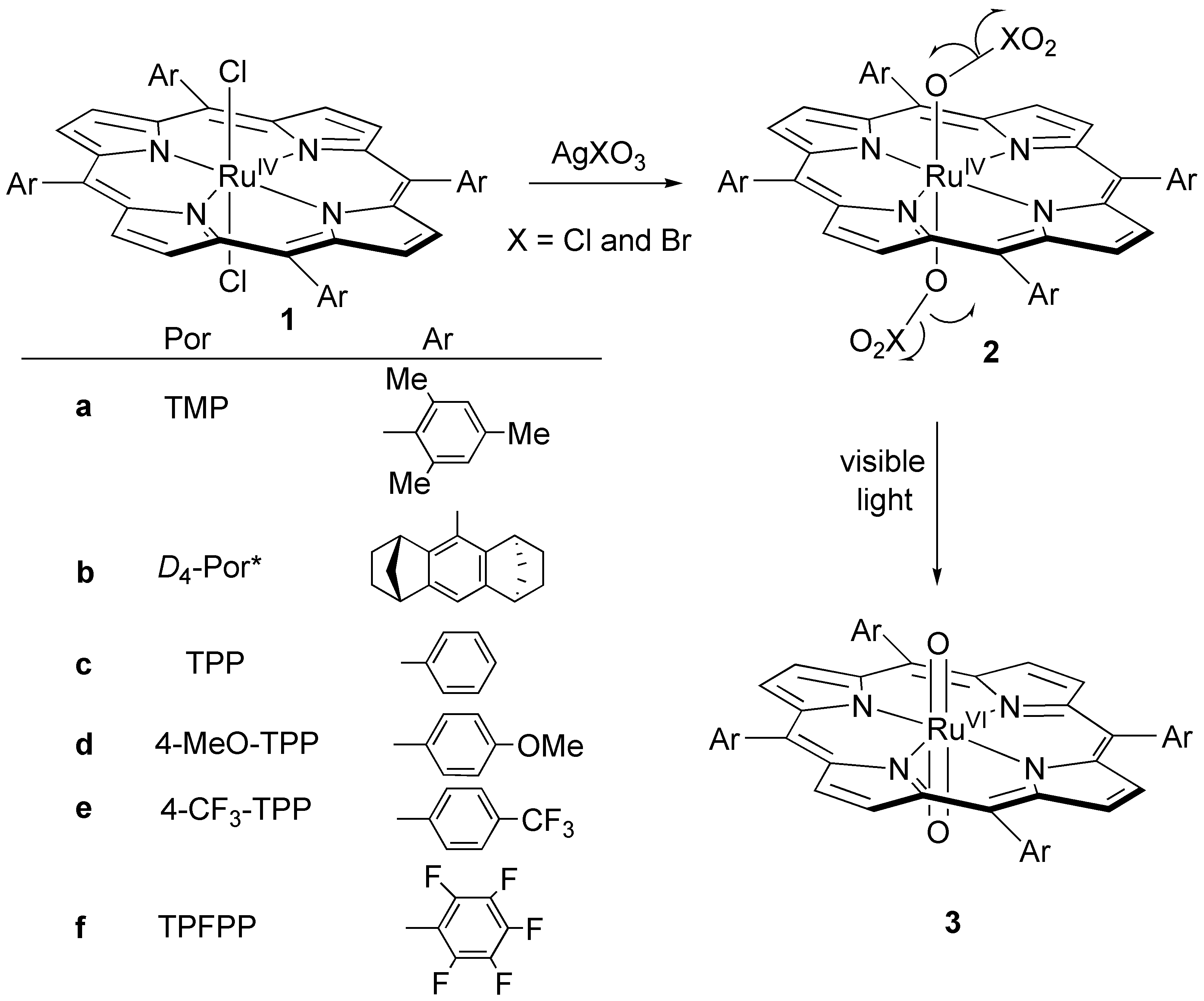
Scheme 1. Photochemical synthesis of trans-dioxoruthenium(VI) porphyrins
We have also discovered that visible light irradiation of the photo-labile dibromate species gave trans-dioxoruthenium(VI) porphyrin (3a) with well-anchored isosbestic points (Figure 1). The photochemical reaction that gave homolytic cleavage of the O-Br bonds is directly the same as that of the ruthenium(IV) dichlorates.

Figure 1. UV-visible spectral change of RuIV(TMP)(BrO3)2 (8 10-6 M) with 5-fold excess of AgBrO3 in anaerobic CH3CN solution upon irradiation with visible light at 22 oC over 80 min.
Kinetic studies of
sulfoxidation reactions by trans-dioxoruthenium(VI) porphyrins
Scheme
2.
Stoichiometric oxidation of thioanisole by trans-dioxoruthenium(VI)
porphyrins
In
kinetic studies, solutions containing the trans-dioxoruthenium(VI)
oxidant were mixed with solutions containing large excesses of sulfide
substrate, and pseudo-first-order rate constants for decay of the ruthenium-oxo
species were measured spectroscopically. The dioxo 3 decayed rapidly in
the presence of the thioanisoles, reacting as fast as 30 seconds. For all
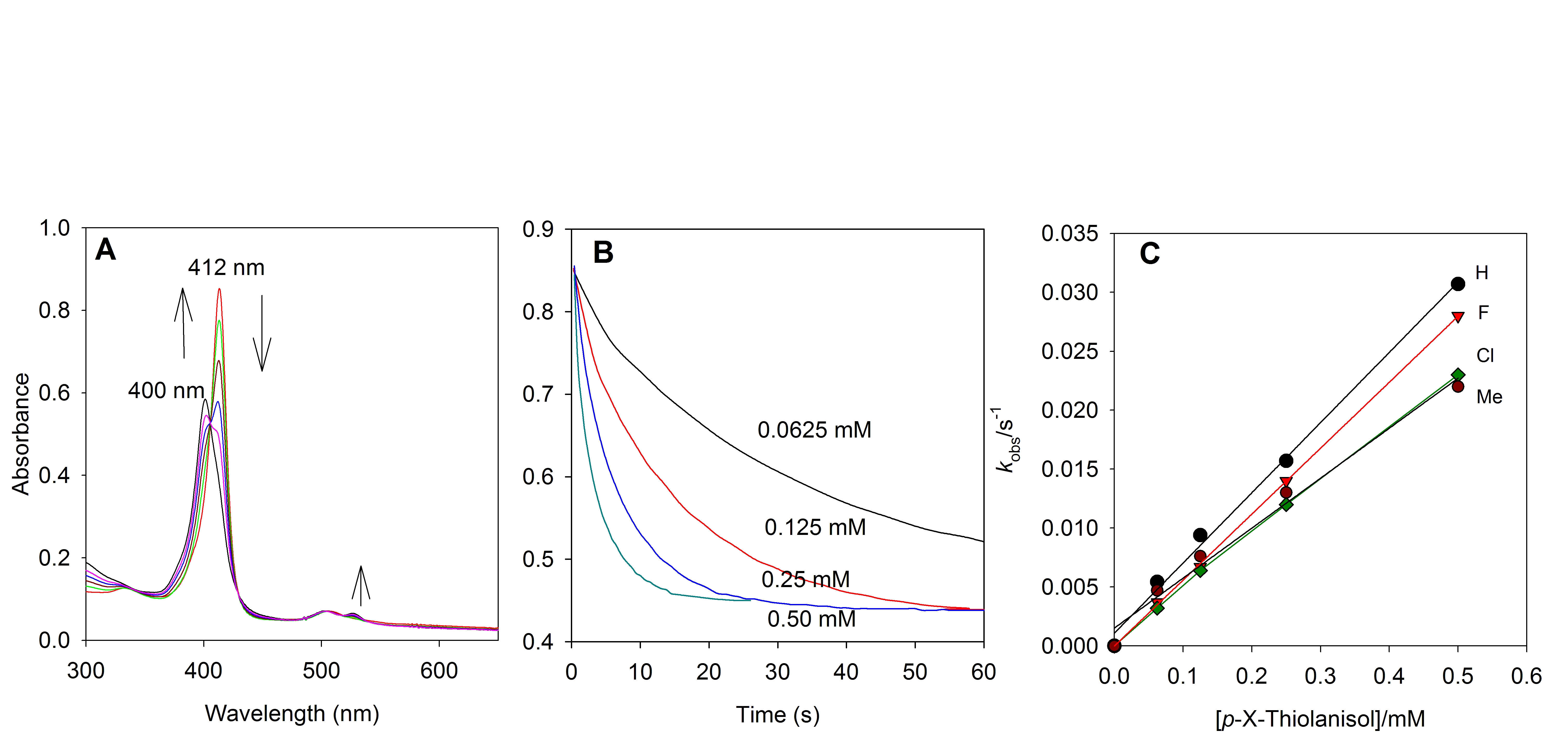
oxo species, we monitored decay of the Soret-band lmax
at 422 nm (3a), 418 nm (3c) and 412 nm (3f). Figure 2
shows typical kinetic results for reactions of TPFPP oxo (3f).
Figure 2. Typical reactions of 3f in CHCl3
at 23 ± 2 oC. (A) Time resolved spectrum over 60 s for reaction of 3f
with 0.25 mM thioanisole. (B) Kinetic traces at 412 nm for reactions of 2c
with thianisole at different concentrations; (C) Plots of the observed
pseudo-first-order rate constants versus the concentration of thioanisole and para-substituted
thioanisoles.
The second-order rate constants for reactions of 3 with a variety of
organic sulfides were summarized in Table 1. For a given oxo species, various
thioanisoles react in a relatively narrow kinetic range; typically the
second-order rate constants for the oxidation of para substituted
thioanisoles with 3f showed little variation (Table 1, entries
7-12). Furthermore, there is no linear correlation between the logkrel
and sp [krel
= k2(substituted thioanisole)/k2(thioanisole)],
implying that no appreciable charge developed on the sulfur during the
oxidation process by dioxoruthenium(VI) species.
Table 1. Rate constants (k2)
for the reactions of [RuVI(Por)O2] (3) with
organic sulfides a
Entry Metal-oxo species Substrate k2 (M-1s-1) 1 2 3 4 5 6 7 8 9 10 11 12 [RuVI(TMP)O2] 3a [RuVI(TPP)O2] 3c [RuVI(TPFPP)O2] 3f thioanisole p-fluorothioanisole p-chlorothioanisole methyl phenyl sulfoxideb thioanisole p-chlorothioanisole thioanisole p-fluorothioanisole p-chlorothioanisole p-methylthioanisole p-methoxythioanisole methyl phenyl sulfoxideb 8.0 ± 0.4 3.0 ± 0.3 8.1 ± 0.5 (3.1 ± 0.4) 10-4 48.0 ± 2.0 38.0 ± 3.0 59.6 ± 2.0 56.0 ± 1.0 42.4 ± 3.0 39.4 ± 2.0 38.0 ± 4.0 (2.6 ± 0.3) 10-2 a Second-order
rate constants in units of M-1s-1 for the
reactions at 22 ± 2oC in CHCl3. The data reported here
are averages of 2-3 runs with a standard deviation of 2s.
b Higher
concentrations (0.2 to 1.0 M) were used for kinetic studies.
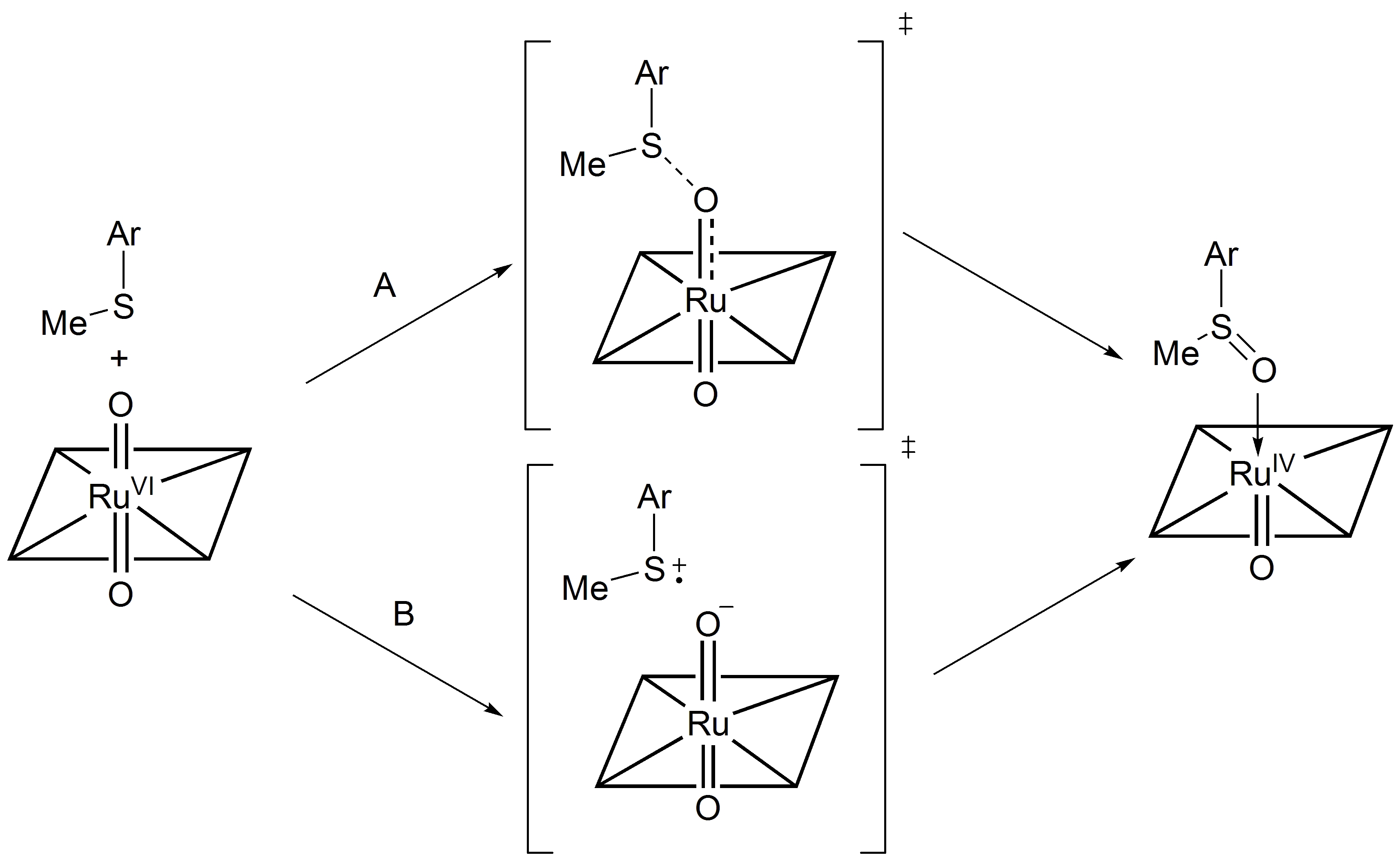
We propose the reactions of trans-dioxoruthenium(VI) compounds
with sulfides proceed via a concerted mechanism where a direct oxygen transfer
occurs from ruthenium to sulfides without the participation of intermediates
(Scheme 3, pathway A). However, the smaller k2 values (entries
10 and 11, Table 1) in the more electron-rich substrates (p-methyl and p-methoxythioanisole)
is indicative of the relatively facile electron transfer from the thioanisoles
without significant S-O bond formation. The contribution of an alternative pathway,
i.e. electron-transfer followed by oxygen transfer (Scheme 3, pathway B), may
be operative in the oxidation of electron-rich substrates.
Scheme
3. Proposed mechanisms for oxygen atom transfer processes from 3 to
thioanisole
Conclusions In summary, we report a new photochemical preparation of trans-dioxoruthenium(VI)
porphyrin complexes in six porphyrin systems. We have also conducted the
kinetic studies of sulfoxidation reactions with these well characterized
metal-oxo species 3. The kinetic results obtained in this study indicate
a concerted oxygen atom transfer and/or electron transfer followed by oxygen
transfer mechanism from oxidant to sulfide.