Reports: G1
47109-G1 Synthesis of Bis-arenes via Oxidative Coupling
The palladium catalyzed oxidative cross-coupling of indoles with arenes is a desirable process because the prefunctionalization of either substrate is not required prior to the coupling reaction, thus allowing for the rapid and green synthesis of arylated indoles. Such processes have been described as key green chemistry research areas by a consortium of leading pharmaceutical companies. Oxidative cross-coupling reactions involving double C-H functionalizations, however, are difficult because the oxidative conditions required often decompose electron-rich indole substrates. Herein, we disclose that oxidative cross-coupling can be achieved and oxidative decomposition can be controlled by careful adjustment of the reaction's pH. Using these mild conditions, the oxidative coupling reaction occurs regioselectively at indole's 2-position. The reaction is amenable to indoles decorated with a wide variety of functional groups, including basic functionalities and protecting groups that can be easily cleaved to provide a diverse library of N-H-2-arylindoles.
In the first year of our project, which was funded by the Petroleum Research Fund, we showed that benzofuran (1) can be oxidatively cross-coupled with benzene to regioselectively form 2-pheynylbenzofuran (2) (Scheme 1, equation 1). The reaction was notable because of its high yield, fast reaction times and use of molecular oxygen as the terminal oxidant. Our initial efforts to apply these optimized conditions to indole substrates were unfruitful. The acidic, oxidizing conditions that were ideal for benzofuran heterocycles rapidly decomposed N-methylindole (3) and other electron-rich substrates (Scheme 1, equation 2).
Subsequently, it was discovered that N-acetylindole and N-pivalylindoles could be oxidatively arylated, and that oxidative decomposition could be repressed, in non-acidic media and anaerobic conditions. Interestingly, both of our conditions allowed for regioselective arylation at either the 2- or 3-position, depending on whether Cu(OAc)2 or AgOAc was used as the terminal oxidant. However, when these conditions were applied to N-alkylindoles, modest yields were observed and were accompanied by a loss of regioselectivity, regardless of the terminal oxidant.
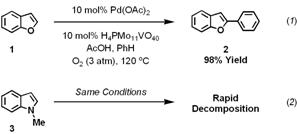
Scheme 1. Limits of Current Oxidative Coupling Technology
In light of this precedent, we
hypothesized that the identity of the inorganic oxidant and the reaction's pH could
be manipulated to tune the reactivity and regioselectivity of the oxidative cross-coupling
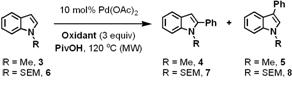
reactions (Table 1). In keeping with our hypothesis, we observed that the yield
of the oxidative coupling of N-methylindole and benzene increased as
modest amounts of pivalic acid were added to the reaction (compare entry 1 with
entry 4). The chemoselectivity of the reaction, favoring 2-arylated indoles,
also increased as the pH of the reaction decreased, but reactions containing
excessive amounts of acid lead to decomposition of both the indole substrate
and products (entry 5). Reactions which substituted Cu(OAc)2 for
AgOAc as the terminal oxidant, demonstrated an inversion in selectivity (entries
6 and 7). This regiochemical phenomenon has been previously observed and was
postulated to be the result of the formation of bimetallic metal acetate
clusters in situ.
Table 1. Discovery and Optimization of Regioselective Oxidative Arylation with Electron-Rich Indoles
|
Entry |
R |
Oxidant |
PivOH |
Yield[a] |
Ratio (4:5)[b] |
|
1 |
Me |
AgOAc |
0 equiv |
10% |
4:1 |
|
2 |
Me |
AgOAc[b] |
0 equiv |
0% |
- |
|
3 |
Me |
AgOAc |
1 equiv |
11% |
6:1 |
|
4 |
Me |
AgOAc |
2.5 equiv |
31% |
4:1 |
|
5 |
Me |
AgOAc |
5 equiv |
26% |
7:1 |
|
6 |
Me |
Cu(OAc)2 |
2.5 equiv |
20% |
1:2 |
|
7 |
Me |
Cu(OAc)2 |
5 equiv |
31% |
1:3 |
|
8 |
SEM[d] |
AgOAc |
2.5 equiv |
69% |
3:1 |
[a] Isolated by flash chromatography (5 + 6). [b] Determined by GC [c] Reaction contained 1 equiv. CsOAc. [d] SEM = 2-trimethylsilyl-ethoxymethyl.
As we were interested in developing a general method for generating a library of diversely functionalized 2-arylindoles, we were encouraged to see that the reaction's yield more than doubled when an indole substrate bearing a readily cleavable N-protecting group was employed (SEM, 6). The optimized reaction conditions contained 2.5 equivalents of pivalic acid and 3 equivalents of AgOAc.
The optimized conditions were also applicable to the oxidative coupling of benzene with indoles containing other N-alkyl groups (Table 2); however, the reactions containing SEM-indole 6 proved to be the highest yielding. N-Tosylindole (12) could also be oxidatively coupled with benzene, and only the 2-phenyl isomer was observed. This result is notable because the tosyl group of 12 significantly diminishes the nucleophilicity of the indole. N-H indole (13) did not oxidatively couple using these conditions, though its oxidative coupling to olefins has been reported.
Table 2. Scope of Oxidative Arylation Reaction
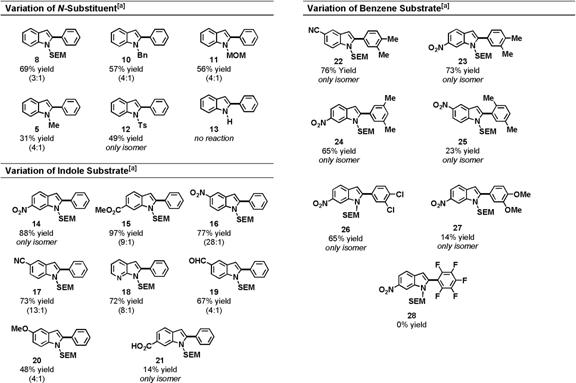
[a] Only the major isomer is shown. The minor isomer is the 3-arylindole. Yields and selectivities were obtained by isolation of both isomers following flash chromatography.
As a general rule, indoles containing electron-withdrawing groups provided higher yields and better regioselectivity for the arylation of the indole's 2-position. Notably, N-SEM-7-azaindole, which could potentially deactivate the catalyst via ligation, coupled with good regioselectivity and high yield (18).
The chemoselective preference for electron-rich arenes is directly opposed to our previous work, in which N-acetylindoles were readily arylated by electron-poor arenes, such as pentafluorobenzene, and were not coupled to electron-rich arenes, such as anisole. These data, which were observed in non-acid media, led us to conclude that the palladation of benzene substrates occurs via a concerted proton abstraction mechanism. In contrast, the reactions described herein likely proceed via a different mechanism, namely electrophilic palladation.
Current efforts in our laboratory are directed towards the application of this technology to the synthesis of high-value molecules such as pharmaceuticals and monomers for conducting polymers. We are very interested in the elucidating the mechanism of these intriguing and important transformations. The project will continue with funding from the National Science Foundation, and new projects involving the oxidative coupling of electron poor arenes and alkanes are beginning in our laboratory. We are sincerely appreciative for the support of the PRF as the aforementioned science could not have been accomplished and numerous scientists could not have been trained without this grant.



