Reports: AC3
47084-AC3 New Homogeneously Catalyzed Dehydrogenation and Hydrogenation of Polar Bonds. Esters and H2 from Alcohols, Hydrogenation of Esters, and Related Reactions
Our accomplishments resulting from this two-year PRF project are listed below:
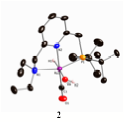
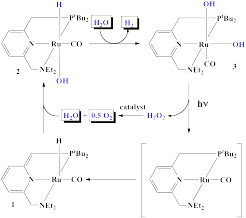
(1) Discovery of an efficient artificial catalyst for the sunlight-driven splitting of water into dioxygen and dihydrogen is a major goal of renewable energy research. In continuation of our work on the activation of alcohol O-H bonds by metal-ligand cooperation, we have studied activation of water. We describe a solution-phase reaction scheme which leads to the stoichiometric liberation of H2 and O2 in consecutive thermal and light-driven steps, mediated by mononuclear, well defined ruthenium complexes. Initial reaction of water at 250C with a dearomatized Ru(II) pincer complex 1 yields the crystallographically characterized monomeric aromatic Ru(II) hydrido-hydroxo complex 2, which on further reaction with water at 1000C releases H2 and forms a cis dihydroxo complex 3. Irradiation of this complex in the 320 – 420 nanometer range liberates O2 and regenerates the starting hydrido-hydroxo Ru(II) complex 2, probably by elimination of hydrogen peroxide, which rapidly disproportionates. Labeling experiments with H217O and H218O show unequivocally that the process of O-O bond formation is intramolecular, establishing a previously elusive fundamental step toward O2-generating homogeneous catalysis.
We believe that our studies indicate a new approach towards a complete cycle for the generation of dihydrogen and dioxygen from water catalyzed by metal complexes, and show that light-induced O-O bond formation need not necessarily involve bimolecular mechanisms, binuclear complexes, and metal oxo intermediates. These studies are described in S. W. Kohl, L. Weiner, L. Schwartsburd, L. Konstantinovski, L. J. W. Shimon, Y. Ben-David, M. Iron and D. Milstein, Science 2009, 324, 74
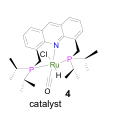
(2) We have discovered a new reaction, namely, the dehydrogenative coupling of primary alcohols to form acetals, i.e. protected aldehydes. The crystallographically characterized complex RuHCl(A-iPr-PNP)(CO) 4 (A-iPr-PNP = 4,5-bis-(di-iso-propylphosphinomethyl)acridine) which bears a non-planar acridine moiety, catalyzes in a neutral medium the transformation of primary alcohols to the corresponding acetals with the liberation of H2. In the presence of base, complex 4 catalyzes the dehydrogenative coupling of alcohols to form esters. Acetal formation may involve hemiacetal dehydration to form an enol ether, followed by alcohol addition to the double bond. This study is reported in C. Gunanathan, L. J. W. Shimon and D. Milstein J. Am. Chem. Soc. 2009, 131, 3146
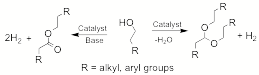
(3) A new pyridine based sulfoxide pincer ligand gives access to cationic rhodium and iridium carbonyl complexes, which can be deprotonated to stable dearomatized rhodium and irdium complexes. As shown by NMR, X-structural analyses and DFT calculations, the pyridine ring is dearomatized and the sulfur atom is also involved in the charge delocalization. This study is reported in T. Schaub, U. Radius, Y. Diskin-Posner, G. Leitus, L.J. W, Shimon and D. Milstein Organometallics, 2008, 27, 1892
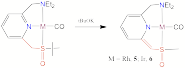
(4) SNS based Rh(I) and Ir(I) complexes are fluxional in solution due to the hemilabile character of the ligand. Oxidative addition of H2 to the Ir(1) complex 7 results in a disulfide-bridged dimeric cis-dihydride complex 8. The carbonyl Rh(I) and Ir(I) complexes 9, which are monomeric in solution, are dimeric in the solid state due to uncommon d8-d8 metal-metal interaction, as shown by X-ray diffraction and by IR spectroscopy. This study is reported in Y. Klerman, E. Ben-Ari, Y. Diskin-Posner, G. Leitus, L.J.W. Shimon, and D. Milstein Dalton Trans., 2008, 3226
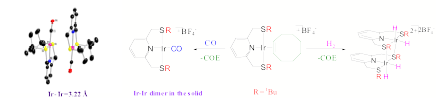 9 9 7 8
9 9 7 8
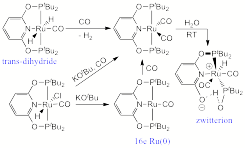
(5) A series of ruthenium complexes based on new phosphinite ligands were prepared, including rare stable trans-dihydride Ru(II) and 16-electron Ru(0) complexes. Treatment of the saturated Ru(0) complexes with water leads to O-P bond cleavage, affording zwitterionic systems. The trans-dihydride complexes show high hydridic reactivity. This study is reported in H. Salem, L. J. W. Shimon, Y. Diskin-posner, G. Leitus, Y. Ben-David and D. Milstein Organometallics, 2009, 28, 4791