

44453-AC1
Development of New Reactions Based on Decarboxylative Metalation
Annual Project Report for Award 44453-AC1
Aim: Develop decarboxylative benzylation of ketone enolates and allylic selenides based on decarboxylative coupling.
Activity: Develop decarboxylative benzylation of ketone enolates
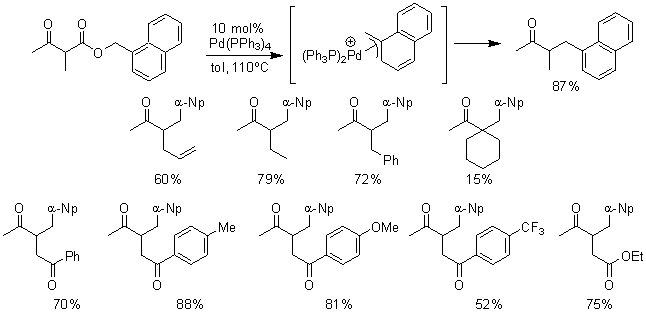
We have developed a new decarboxylative benzylation of ketone enolates. Previously, we have shown that ketone enolates can be generated under mild conditions by palladium-catalyzed decarboxylation of b-keto carboxylates. This reaction relied on the insertion of palladium into the C-O bond of an allylic ester. Now we are addressing the much more difficult challenge of inserting palladium into the C-O bond of benzylic esters and coupling the resulting p-benzyl complex with ketone enolates. In doing so, we have discovered that simple Pd(0) catalysts can effect decarboxylative benzylation of ketone enolates. To begin we have utilized 2-naphthyl b-ketoesters as reactants to explore their chemistry. Treatment of a simple b-ketoester with Pd(PPh3)4 in toluene at reflux produced the product benzylated ketone in 87% yield. Importantly, no isomerization of the intermediate enolate takes place. A variety of simple alkyl substituents are also compatible with the reactions conditions. Most noteworthy is the fact that substituents with other, more acidic, ketones and esters are compatible; no isomerization of the enolate generated via decarboxylation to the more stable enolate is observed.
Activity: Decarboxylative coupling of allylic selenides.
We have previously developed a straightforward route to allylic selenocarbonates via decarboxylative selenylation. Here, treatment of the selenoformate with catalytic Pd(PPh3)4 indeed provided the allyl selenides in good yield. This was particularly promising since other methods for palladium-catalyzed selenation failed to give any of this product. Our hypothesis is that, while nucleophilic selenides often poision palladium catalysts, the selenocarbonate selenium atom is less nucleophilic, thus the palladium catalyst does not readily poison. Having done that, we have turned our attention to developing an asymmetric allylic selenylation reaction. We have now conducted exhaustive screening of ligands and have identified the naphthyl Trost ligand as the best ligand for asymmetric decarboxylative selenylation reactions. The results of some of these experiments are shown below. It is particularly interesting to note that the highest ee's are often obtained for reactions that proceed to <60% conversion.

The correlation of low ee's with higher conversions suggested that kinetic resolution of the substrate may be taking place. Thus, we have tested to see whether the reaction is occurring by kinetic resolution, and indeed our experiments have proven that we are effecting the kinetic resolution of the selenocarbonates. The resolution is quite efficient. For example, for substrates like 4g, the selectivity factor for resolution can be as high as 80.

With the ability to synthesize enantioenriched allyl selenides, it seemed that the transformation of the allyl selenides to the corresponding allylic chlorides and amines via a [2,3]-sigmatropic rearrangement to give enantioenriched allylic amines and chlorides would be also be synthetically useful. For example, allylic amines can be obtained by oxidation with chloramine T. However, the yield of this reaction is low (ca. 40%), so optimization of the reaction conditions is still necessary. A better transformation is the treatment of allyl selenides with N-chlorosuccinimide (NCS) and anilines which gives rise to optically active allylic amines with high stereochemical fidelity. Perhaps even more exciting, we have found that chlorination with NCS alone provides the allyl chlorides with high conservation of enantiomeric excess. This is noteworthy because we are unaware of any other method that can produce such highly enantioenriched allylic chlorides.
