

44683-G1
Guanidinium Catalysts for the Asymmetric Addition of Nucleophiles to N-Acylhydrazones
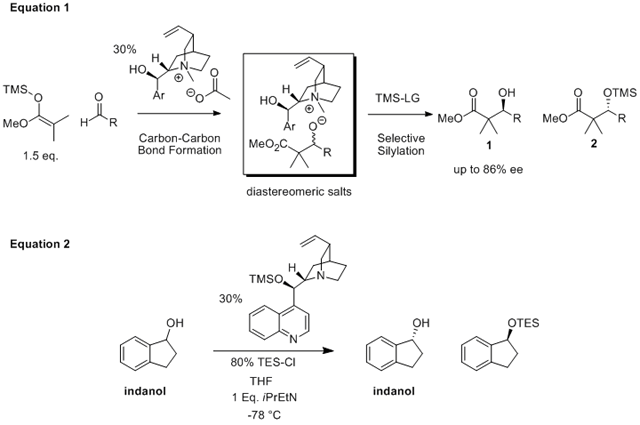
The goal of the original project was the development of reactions promoted by organocatalyts. To date our efforts have focused on the use of nucleophilic catalysts to catalyze reactions through a Lewis base mechanism. This project involved the kinetic resolution of secondary alcohols which is an important transformation for the preparation of chiral building blocks. While there are a number of ways to selectively derivatize one alcohol enantiomer over another, the use of a silyl group has been virtually ignored. Therefore we are working on the asymmetric silylation of secondary alcohols via two different approaches: differentiation of diastereomeric salts (Eq. 1) and chiral nucleophilic catalysts (Eq. 2). Both approaches have shown promising results in achieving enantioselectivity.
SHAPE \* MERGEFORMAT
|
Chiral Cation Silylations
We have explored the Mukaiyama aldol reaction, investigating the fact that this transformation is actually a combination of two reactions: a carbon-carbon bond forming reaction and a protection step (silylation of the newly formed alkoxide). We have identified reaction conditions where the enantioselectivity of our Mukaiyama aldol reaction arises exclusively from the silylation step. In this process, the first step is not enantioselective, yielding racemic aldolate, which is then enantioselectively silylated in the second step (Eq. 1). Thus this is an example of a kinetic resolution employing a silyl protecting group. (Submitted, Tetrahedron Lett. 2008)
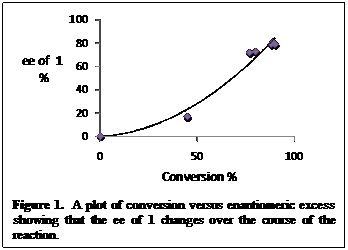
We investigated the mechanism of this reaction in more detail. The conversion of the starting material into product and the enantiomeric excess of the reaction were monitored over the course of the reaction. Figure 1 shows that enantioselectivity of the alcohol varies over the course of the reaction and increases with increasing conversion which is consistent with a kinetic resolution process. We also looked at the enantiomeric excess of both the left over starting material and the product of the reaction. These two compounds should have opposite chirality which was the case. The compounds from our system were enriched in the opposite enantiomers, the alcohol product 68% (S) and the silylated product 8% (R).
SHAPE \* MERGEFORMAT
Figure 1. A plot of conversion versus enantiomeric excess showing that the ee of 1 changes over the course of the reaction.
|
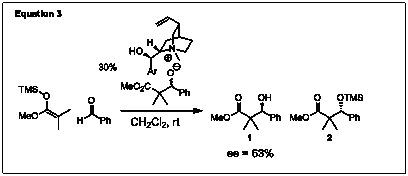
Another approach to investigating the mechanism that was employed consisted of showing that the diastereomeric salts could generate enantiomeric excess when introduced as the catalyst at the start of the reaction. The diastereomeric salts were preformed and subjected to a mixture of silyl ketene acetal and aldehyde. The reaction progressed as expected forming products 1 and 2, and an ee of 63% was determined for 1 (Eq. 3). This showed that the diasteromeric salts could catalyze carbon-carbon bond formation while selectively silylating one enantiomer of the alkoxide resulting in a kinetic resolution.
SHAPE \* MERGEFORMAT
|
Nucleophilic Enantioselective Silylations.
On the last report, we summarized our work thus far in developing a nucleophilic catalyzed asymmetric silylation reaction. We have continued to develop this project through investigation of various aspects of the reaction. We have explored the effect of the R groups on the silyl, the silyl leaving group, the base, the catalyst, and the solvent. We have found conditions that selectivity can be obtained, and the future work will consist of looking at different substrates to see how sterics and electronics affect the reaction.
In conclusion, we have made a lot of progress over the last year, developing two asymmetric silylation reactions. This grant has helped my group tremendously through providing funding to start a new project. The award aided in funding a graduate student on a research assistantship as well as purchase supplies and chemicals. We have obtained enough data to submit one paper and we are working towards a second paper. We would like to thank the donors of the ACS-PRF foundation for their support.