

44783-G1
Lewis Acid Activation of Dioxiranes for Aliphatic C-H Bond Oxidation
Overview. The interaction between a drug and its target protein is typically mediated by hydrogen bonds between oxygen- and nitrogen-containing functional groups on the drug and receptors on the protein. The specificity and strength of this interaction is dictated by the arrangement of the drug's functional groups in space. The ultimate starting materials for most synthetic organic compounds, however, are simple petrochemical hydrocarbons (e.g., alkanes, alkenes, and arenes) that do not bear functional groups, cannot hydrogen bond, and are not stereochemically well-defined. Thus, a fundamental challenge in synthetic organic chemistry is the development of powerful, general methods for the selective installation of functional groups onto unfunctionalized hydrocarbon substrates with precise control over the position and spatial arrangement of these new functionalities.
Initially, we attempted to address this important problem by examining the ability of transition metal catalysts to control the reactions of dioxiranes with hydrocarbons. However, the instability of dioxiranes towards isolation and the aqueous conditions under which they are generated impeded our early investigations. We have elected instead to examine the reactions of structurally analogous oxaziridines, which perform many of the same reactions as dioxiranes. In contrast to dioxiranes, however, oxaziridines are stable towards isolation, and the ability to investigate the chemistry of oxaziridines under controlled conditions encouraged us to examine their reactivity as a model system.
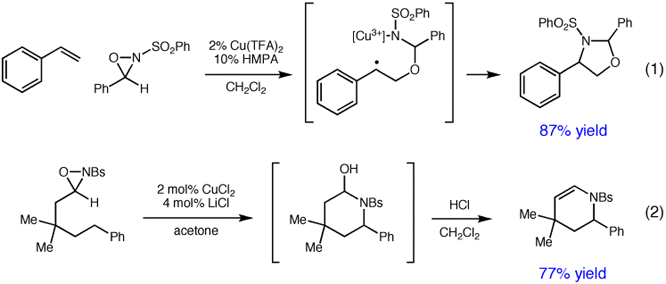
Copper-catalyzed reactions of oxaziridines. In our last annual report to the PRF, we described our discovery of a new copper(II)-catalyzed reaction in which the nitrogen and oxygen atoms of oxaziridines are added across the carbon-carbon double bonds of styrenes in a highly regioselective manner. This constitutes a formal aminohydroxylation reaction, allowing efficient access to 1,2-aminoalcohols in good yields from a variety of electronically varied and substituted styrenes. An important feature of this new method is the use of an inexpensive and relatively innocuous copper catalyst, in contrast to the toxic and expensive osmium catalyst required for the Sharpless aminohydroxylation that is the conventional method for this oxidative transformation.
Our research on this project over the past year has been driven by our desire to understand this novel reactivity. From a variety of studies, we have developed the mechanistic proposal shown in eq 1. The oxaziridine becomes activated upon coordination to copper and undergoes homolytic attack on the styrenic olefin. Ring closure by attack of the copper(III) sulfonamide on the resulting benzylic radical produces the regioisomer of the product that we observe.
This mechanism is consistent with the observation that primary aliphatic olefins, which would produce less stabilized secondary radicals, react inefficiently under these conditions. On the other hand, we speculated that other substrates that would stabilize the radical intermediate would be excellent substrates for aminohydroxylation. Indeed, a variety of electron-rich substrates, including vinyl ethers, dienes, and allyl silanes, are successfully aminohydroxylated using this new method. We also recognized that this mechanism requires the intermediacy of a highly oxidized copper(III) sulfonamide. It follows from this analysis that modifications that would stabilize this oxidation state might also increase the rate of the aminohydroxylation. A survey of more electron-rich copper catalysts showed that anionic halocuprate(III) complexes such as Bu4N+CuCl3- are dramatically more efficient catalysts for aminohydroxylation. A manuscript detailing this result and the full details of our mechanistic investigations is currently in preparation.
More recently, we have investigated the use of the more reactive anionic CuCl3- catalysts in oxidative functionalizations of alkanes, which are more challenging substrates for oxidation. We discovered that treatment of 3-alkyl oxaziridines with a copper(II) catalyst results in an efficient, highly regioselective intramolecular amination of the alkyl side chain. After acid-catalyzed dehydration, a variety of substituted piperidine derivatives can be accessed in high yields (eq 2). The reaction efficiency is highest when the C-H bond is benzylic; however, fully aliphatic linear alkyl oxaziridines also produce piperidines as the only observed product of oxidative cyclization.
This discovery is notable for a number of reasons. First, it is a novel approach to amination of C-H bonds that is conceptually and mechanistically distinct from the metal-nitrene insertion methods that have been reported to date. Second, this reaction is the first example of formal nitrogen atom transfer from an N-sulfonyl oxaziridine, which demonstrates that fundamentally new reactions of oxaziridines can be discovered in the presence of transition metal catalysts. Finally, as we proposed in our original application to the PRF, we have demonstrated that electrophilic catalysts can indeed promote the intramolecular oxidative functionalization reactions of this class of oxidizing heterocycles. This strategy promises to continue to be a rich platform for reaction discovery for us and other researchers in the future.