45277-B1
Investigation of the Butyllithium-Induced Sigmatropic Rearrangement: Intramolecular Cyclization Reaction of Allyl-Dichlorovinyl Ethers
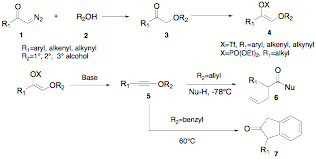
The sigmatropic rearrangement of in-situ generated allyl-alkynyl ethers is a powerful carbon-carbon bond forming process that is stereospecific and takes place instantaneously at low temperatures. Our original formulation for this reaction involved the treatment of allyl-1,1-dichlorovinyl ethers with n-butyllithium at -78°C, followed by reaction quench with an alcohol, to afford g,d-unsaturated esters in high yields. Although efficient and stable precursors of allyl-alkynyl ethers, allyl-1,1-dichlorovinyl ethers must be prepared by reaction of allyl formate esters with the toxic reagent combination triphenylphosphine-carbon tetrachloride; furthermore, the use of the strong nucleophilic base (n-BuLi) to generate the reactive intermediate in this reaction may be viewed as a potential limitation. Through the support of this ACS-PRF grant, we sought both to overcome the drawbacks of this process, as well as to expand its utility to include the formation of up to three new carbon-carbon bonds in a single-step process. With the combined efforts of undergraduate students Juan Sosa and Armen Tudjarian, we have discovered a facile and broadly applicable methodology for the preparation of a diverse range of alkynyl ethers. Thus, combination of a-diazoketones 1 and alcohols 2 in the presence of an indium triflate catalyst leads to a-alkoxy ketones 3 in high yields; formation of the enol triflate (for aryl, alkenyl, and alkynyl ketones) or enol phosphate (for alkyl ketones) is followed by t-BuOK induced E2-elimination to form the substituted alkynyl ethers 5. This method does not require the use of toxic reagents and uses a non-nucleophilic base for alkynyl ether generation. Furthermore, when allylic alcohols are used in this procedure, the intermediate allyl alkynyl ethers undergo rapid [3,3]-sigmatropic rerrangement at temperatures as low as -85°C to furnish an allyl ketene intermediate; subsequent addition of 1°, 2°, 3° alcohols or amines lead to g,d-unsaturated esters or amides 6 in high yields. Similarly prepared benzyl alkynyl ethers are stable at room temperature but rapidly rearranged at 60°C in toluene to furnish 2-substituted indanones 7 (Scheme 1). This work has significantly expanded the practicality and utility of the allyl-alkynyl ether rearrangement reaction. Armen Tudjarian has decided to continue his studies as a masters' student in my laboratory; Juan Sosa is also continuing his work in my laboratory and is considering going to graduate school in chemistry.
Scheme 1 Our
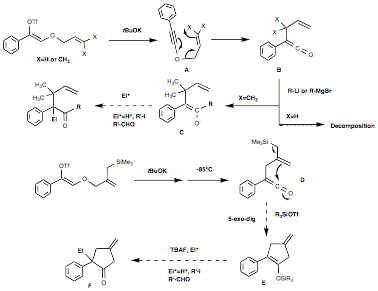
current focus is on trapping the ketene intermediate (formed upon
[3,3]-sigmatropic rearrangement) with carbon nucleophiles to form substituted
ketone products. Treatment of the ketene intermediate at -85°C with an
organolithium or Grignard reagent is expected to give rise to a ketone enolate
capable of reacting with an externally added carbon electrophile. We have
discovered that this approach is feasible only if deprotonation of the allyl
ketene intermediate is precluded; thus prenol-derived allyl-alkynyl ethers (A, X=CH3,
scheme 2) are ideal substrates for exploring the utility of this
transformation. If successful, this approach will provide a new regioselective
ketone synthesis, in which contiguous quaternary centers are formed in a single
synthetic operation.
Scheme 2. Finally,
we are also currently investigating the trapping of the ketene intermediate in
an intramolecular fashion to form substituted cyclopentanones. By coupling
2-(trimethylsilylmethyl)allyl alcohols to a-diazoketones and subsequent alkynyl ether formation
and rearrangement, we have obtained ketenes containing the nucleophilic
allylsilane moiety capable of undergoing 5-exo-dig cyclization (D, scheme 2). At -85°C or -78°C, reaction does not
take place, and thus a Lewis acid promoter must be added to facilitate
cyclization. We are currently exploring the use of silyl triflate additives for
this reaction, which would furnish silyl enol ether products E capable of undergoing a further reaction with an
electrophile in the presence of fluoride ion. Again, if successful, this
approach will provide a new regioselective synthesis of substituted
cyclopentanones, which are key compounds useful for the construction of the
complex ring systems present in a variety of natural products. This
research has opened up a new area of chemistry for exploration with exciting
possibilities for the development of tandem carbon-carbon bond-forming
reactions. Furthermore, we believe that the [3,3] sigmatropic processes we are
studying may be considered as both environmentally
friendly and atom economical; indeed the development of efficient and benign
chemical reactions are key considerations for chemists both in industry and
academia today. Finally, we are proud to have our research students deeply
involved in this exciting area of cutting-edge scientific research, and we are
confident that they will reap the benefits of this practical experience in
their future careers.