
Back to Table of Contents
45648-AC3
Lewis Acid Chemistry at the Edge of Ferrocene
Frieder Jäkle, Rutgers, the State University of New Jersey (Newark)
Bidentate
Lewis acids have attracted much recent interest due to their superior
performance in diverse areas ranging from Lewis acid catalysis of
organic and
organometallic reactions, activation of catalysts in olefin
polymerization,
selective and efficient molecular recognition of anions and neutral
nucleophiles, to new electronically interesting materials. The purpose
of the
research under this grant was to explore the chemistry of ferrocenes
with two
adjacent Lewis acid centers attached to one of the cyclopentadienyl
rings.
While the more easily accessible 1,1Õ-dimetalated ferrocenes
have been widely
studied in the past, the chemistry of the related 1,2-difunctionalized
ferrocenes is virtually unexplored. The latter are of particular
interest since
they represent three-dimensional, redox-active analogs of the
well-established
and highly useful bidentate Lewis acids with phenylene and naphthalene
backbones. During
the current grant period we have (A)
Succeeded
in the chiral resolution of heteronuclear bidentate Lewis acids that
feature
Lewis acidic organotin and organoboron moieties adjacent to one another
at the
edge of ferrocene and pursued their application in stereoselective
organic
transformations,(B)
Studied
the electronic structure and binding behavior of diboradiferrocenes, in
which
two adjacent Lewis acidic organoboron moieties lead to a doubly-bridged
diferrocene system; a new route to these bifunctional Lewis acids that
allows
us to optimize the electronic structure through variation of the
exocyclic boron
substituents was developed.(A) Chiral
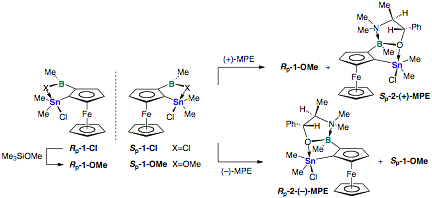
Resolution of Heteronuclear Bidentate Lewis Acids. We first converted
the bidentate
Lewis acid 1-Cl to the
methoxy
derivative 1,2-Fc(BMeOMe)(SnMe2Cl) (1-OMe) and then treated the latter with 0.5
equiv of (1R,2R)-(–)-N-methyl pseudoephedrine ((–)-MPE) and (1S,2S)-(+)-N-methyl
pseudoephedrine ((+)-MPE),
respectively (Scheme 1). The chelate complexes Rp-2-(–)-MPE and Sp-2-(+)-MPE were isolated as
yellow solids
with >97% de. The remaining unreacted isomers Rp-1-OMe
and Sp-1-OMe,
respectively, were subsequently converted back to the now
enantiomerically pure
bidentate Lewis acid Rp-1-Cl
and Sp-1-Cl.
Scheme 1. Chiral resolution of the bidentate Lewis
acid 1-Cl by
complexation with N-methyl
pseudoephedrine (MPE).
The new planar chiral
organoboranes were then converted to the allyl derivatives, which in
turn were
successfully employed in the stereoselective allylation of ketones
(upto 80%
ee).
(B)
Binding
studies on diboradiferrocenes and tuning of the electronic structure. We have developed a
new and more
versatile route to diboradiferrocenes starting from
1,2-bis(chloromercurio)ferrocenes. Conversion to the chloro-substituted
diboracycle (fc2B2Cl2) was achieved by
treatment with BCl3 at elevated temperature. The latter
serves as a
universal precursor to a variety of new diboradiferrocenes. Of
particular
interest to us was the possibility to tune the electronic structure of
the
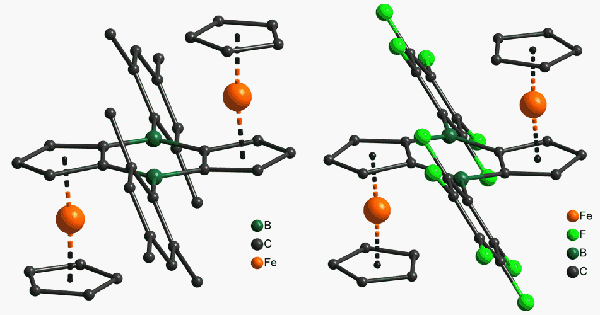
bridge by variation of the exocyclic substituent on boron. The crystal
structures of two new diboraferrocenes with mesityl (2-Mes) and electron-withdrawing
pentafluorophenyl (2-Pf)
groups are shown in Figure 1.
Figure 1. Molecular structures of 2-Mes and 2-Pf.
Importantly,
studies on the electronic structure of 2-Mes and 2-Pf by UV-visible spectroscopy and cyclic
voltammetry suggest a strong
impact of the exocyclic substituent on the degree of electronic
communication
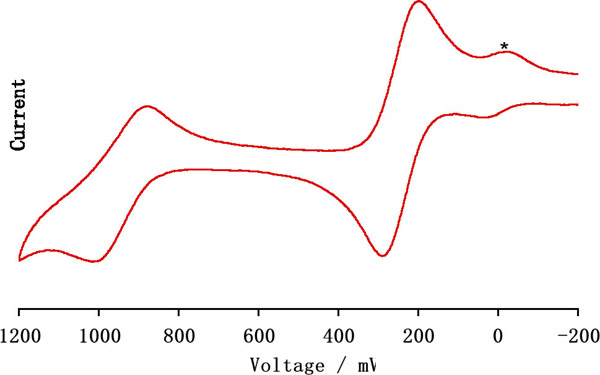
between the ferrocene moieties. Specifically, CV studies on 2-Pf revealed an exceptionally large redox
coupling
between the first and second oxidation (Figure 2), to our knowledge the
largest
reported to date for a heteroatom-bridged diferrocene species.Figure 2. Cyclic voltammogram of 2-Pf reported vs. FcH/FcH+;
* denotes a trace of ferrocene.
Back to top
