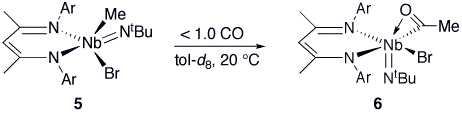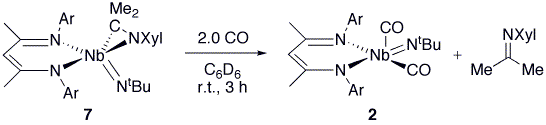47249-AC3
Fundamental Chemistry of New Group 5 Metal Imido and Bis-Imido Complexes
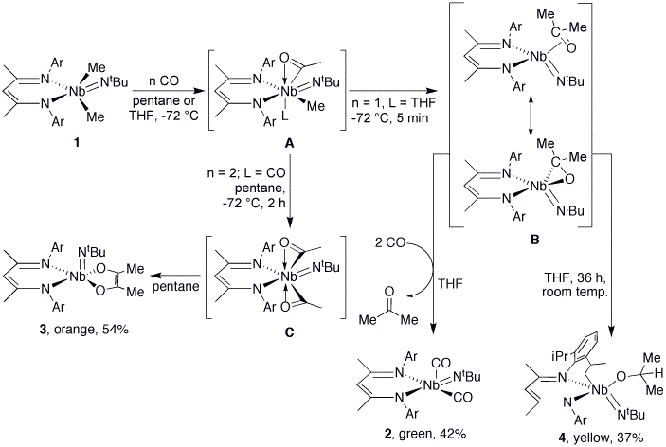
The clean, efficient utilization of carbon monoxide as a C1 source continues to represent a significant chemical challenge. Stoichiometric reactions of carbon monoxide with simple alkyl and dialkylmetal complexes usually lead cleanly to carbonylation products. Through this stoichiometric chemistry, many of the classic primary steps in homogeneous late metal-mediated catalytic organometallic reactions, such as migratory CO insertion, have been identified.1 Herein we describe the carbonylation reaction of the dimethylniobium complex (BDI)Me2Nb(NtBu) (1, BDI = beta-diketiminate), in which the relative amounts of an unusually wide range of products change dramatically in response to very modest changes in reaction conditions. Four different stable products, in which CO insertion takes place coupled with metal reduction, enediolate formation and C-H activation, are formed in these transformations. By careful adjustment of reaction conditions, the majority of these materials have been isolated and fully characterized.
One product was identified as the Nb(III) dicarbonyl adduct (BDI)(CO)2Nb(NtBu) (2, 42% isolated yield, Scheme 1), which can be considered to result from transfer of both methyl groups to one CO, followed by the displacement of acetone by two molecules of CO (νCO (2) = 1988, 1893 cm-1). The formation of acetone was confirmed analytically.
Scheme 1
The outcome of the reaction of 1 with CO may be altered dramatically by using pentane
as the solvent in place of THF. In
so doing, a second product may be isolated, whose analytical data are
consistent with its formulation as the enediolate species (BDIiPr)(Me2C2O2)Nb(NtBu) (3,
54% yield, Scheme 1). A third product from the reaction of CO with 1 is obtained by slowly introducing 1.0 equiv of CO
into a solution of 1 in THF,
yielding {ArNC(Me)CHC(Me)N[2-(CHMeCH2)-6-iPr-C6H3]}(Me2HCO)Nb(NtBu) (4,
Scheme 1). Compound 4 appears to
form from a net process of transfer of both methyl groups to the CO carbon and
the formal addition of a ligand C-H bond across the Nb-O-C linkage. Compounds 2-4 presumably originate from an initial mono-acyl
complex (A, Scheme 1). Support for the formulation of A as a monoacyl is provided by treating a monoalkylated
complex (BDI)MeBrNb(NtBu)
(5) with 13CO. In this
case a single product is observed with a 13C NMR resonance at 292
ppm, attributable to the species (BDI)Br(Me13CO)Nb(NtBu) (6,
Scheme 2). The 1H-coupled 13C NMR spectrum reveals the
signal to be a pseudo-quartet (93Nb, 100%, I = 9/2) with 2JC-H = 6.5 Hz, corresponding to a doublet
with the same coupling constant in the 1H NMR spectrum at 2.56 ppm. Scheme 2
The factor determining the product distribution
appears to be the coordinating ability of the solvent. In pentane, free CO
coordinates to the metal center and undergoes insertion into the remaining
Nb-Me bond to form a diacyl complex, leading to the enediolate, 3. In THF, however, the solvent occupies a coordination
site preferentially over CO, allowing methyl transfer to the acyl group to
occur on warming. This second methyl group transfer amounts to a formal
reduction of Nb(V) to Nb(III). Displacement of the coordinated acetone with two
equivalents of CO then leads to the formation of 2. The formation of 4 could conceivably proceed through a number of
pathways. The formation of 4-d6 from (BDI)(CD3)2Nb(NtBu) (1-d6) revealed
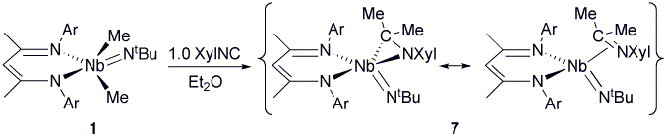
that the two diastereotopic deuterium-labeled methyl groups remained intact. To gain further insight into the possible intermediacy
of complex B, the reaction of 1 with isocyanides, an isoelectronic variant of CO, was
investigated, yielding (BDI)(η2-XylNCMe2)Nb(NtBu) (7,
Scheme 3). While an X-ray diffraction study indicates the product of the
reaction to be a Nb(V) azaniobacyclopropane, reactivity studies imply
assignment of the oxidation state to be more complicated. Scheme 3
On reacting 7 with 2.0 equiv of CO in C6D6 at room
temperature, 2 grows in cleanly
and quantitatively with concomitant formation of free ketimine XylN=CMe2
(Scheme 4), indicating that 7 may
react through a mechanism that involves the metal center exhibiting Nb(III)
character. Scheme 4
The contrast between the oxidation states assigned to
reactions involving 7 and the
ground state of 7 (XRD) allow for
informed speculation as to the mechanism leading to 4. While intermediate B would yield 2 by displacement of acetone with 2.0 equiv of CO, B may also be described as a Nb(V)
oxaniobacyclopropane, which, in the absence of excess CO, would ring-open (due
to strain within in the three-membered metallacycle) by way of an
intramolecular C-H bond activation to yield 4. This behavior would reflect the dual Nb(III)/Nb(V)
character exhibited by the electronically and structurally analogous ketimine
complex 7. These results indicate
an ability to dial in a specific product mixture based on very subtle
modifications to the reaction conditions – a technique particularly
useful in transforming CO into various organic species. (1) For relevant literature citations throughout this summary, see
those given within the communication listed on the associated bibliography.